 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5ahz: Bromide-bound form of Halorhodopsin from Halobacterium salinarum ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5ahz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Bromide-bound form of Halorhodopsin from Halobacterium salinarum in a new rhombohedral crystal form | ||||||
 Components Components | HALORHODOPSIN | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / TRANSPORT PROTEIN / HALOPHILIC ARCHAEA / ARCHAEAL RHODOPSIN / LIGHT DRIVEN ION PUMP / RETINAL PROTEIN / ION TRANSMEMBRANE TRANSPORT | ||||||
| Function / homology |  Function and homology information Function and homology informationphototransduction / photoreceptor activity / monoatomic ion channel activity / plasma membrane Similarity search - Function | ||||||
| Biological species |  HALOBACTERIUM SALINARUM (Halophile) HALOBACTERIUM SALINARUM (Halophile) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.45 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.45 Å | ||||||
 Authors Authors | Schreiner, M. / Schlesinger, R. / Heberle, J. / Niemann, H.H. | ||||||
 Citation Citation | Journal: J.Struct.Biol. / Year: 2015 Title: Structure of Halorhodopsin from Halobacterium Salinarum in a New Crystal Form that Imposes Little Restraint on the E-F Loop. Authors: Schreiner, M. / Schlesinger, R. / Heberle, J. / Niemann, H.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5ahz.cif.gz | 61.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5ahz.ent.gz | 43.8 KB | Display | PDB format |
| PDBx/mmJSON format | 5ahz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ah/5ahzftp://data.pdbj.org/pub/pdb/validation_reports/ah/5ahz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5ahySC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27945.709 Da / Num. of mol.: 1 / Fragment: RESIDUES 20-274 Source method: isolated from a genetically manipulated source Details: IMINE BOND (SCHIFF BASE) BETWEEN NZ OF LYS242 AND C15 OF THE RETINAL LIGAND Source: (gene. exp.) HALOBACTERIUM SALINARUM (Halophile) / Strain: L33 / Production host: HALOBACTERIUM SALINARUM (Halophile) / Strain (production host): L33 / References: UniProt: B0R2U4 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-RET /   Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O | ||||||||||
| #3: Chemical |   Mass: 79.904 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Br Mass: 79.904 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Br#4: Sugar | ChemComp-BOG / | 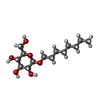  Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 1 Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C14H28O6 / Comment: detergent*YM #5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 24 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 24 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Nonpolymer details | RETINAL (RET): RETINAL IS COVALENTLY | Sequence details | AMINO ACIDS 1-19 ARE MISSING (CLEAVED SIGNAL PEPTIDE), C- TERMINAL HEXAHISTID | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.78 Å3/Da / Density % sol: 55.82 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 296 K / Method: vapor diffusion Details: RESERVOIR SOLUTION: 100 MM CITRATE PH 8, 150 MM NABR, 2.3 M (NH4)2SO4. VAPOR DIFFUSION AT 296 K WITH DROP RATIO OF 1.2 TO 0.8 UL PROTEIN TO RESERVOIR. PROTEIN CONCENTRATION 7 MG/ML. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 0.8726 / Beamline: ID23-2 / Wavelength: 0.8726 |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Jul 22, 2013 / Details: PT COATED SI MIRROR |
| Radiation | Monochromator: CRYSTAL SI (111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8726 Å / Relative weight: 1 |
| Reflection | Resolution: 2.45→42.8 Å / Num. obs: 10814 / % possible obs: 100 % / Observed criterion σ(I): -3 / Redundancy: 6.2 % / Biso Wilson estimate: 40.53 Å2 / Rmerge(I) obs: 0.12 / Net I/σ(I): 14.3 |
| Reflection shell | Resolution: 2.45→2.54 Å / Redundancy: 6.3 % / Rmerge(I) obs: 1.06 / Mean I/σ(I) obs: 1.98 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: ISOMORPHOUS STRUCTURE OF CHLORIDE-BOUND HALORHODOPSIN, PDB ID 5AHY Resolution: 2.45→42.836 Å / SU ML: 0.32 / σ(F): 1.35 / Phase error: 26.8 / Stereochemistry target values: ML Details: RESIDUES 263-274 AS WELL AS THE C-TERMINAL HEXAHISTIDINE-TAG ARE DISORDERED
| |||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 41.1 Å2 | |||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.45→42.836 Å
| |||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
