Entry Database : PDB / ID : 4ywyTitle Crystal Structure of double mutant Y115E Y117E human Glutaminyl Cyclase in complex with inhibitor PBD-150 Glutaminyl-peptide cyclotransferase Keywords / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 1.95 Å Authors Di Pisa, F. / Pozzi, C. / Benvenuti, M. / Mangani, S. Journal : Acta Crystallogr.,Sect.F / Year : 2015Title : The soluble Y115E-Y117E variant of human glutaminyl cyclase is a valid target for X-ray and NMR screening of inhibitors against Alzheimer disease.Authors : DiPisa, F. / Pozzi, C. / Benvenuti, M. / Andreini, M. / Marconi, G. / Mangani, S. History Deposition Mar 21, 2015 Deposition site / Processing site Revision 1.0 Aug 5, 2015 Provider / Type Revision 1.1 Aug 12, 2015 Group Revision 1.2 Aug 19, 2015 Group Revision 1.3 Jan 10, 2024 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / struct_conn Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj
 Assembly
Assembly





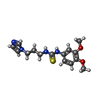




 Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn Mass: 320.410 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C15H20N4O2S
Mass: 320.410 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C15H20N4O2S Mass: 62.068 Da / Num. of mol.: 20 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 20 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Sample preparation
Sample preparation / Beamline: I04-1 / Wavelength: 0.979 Å
/ Beamline: I04-1 / Wavelength: 0.979 Å Processing
Processing