 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4v08: Inhibited dimeric pseudorabies virus protease pUL26N at 2 A resolution -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4v08 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Inhibited dimeric pseudorabies virus protease pUL26N at 2 A resolution | |||||||||
 Components Components | UL26 | |||||||||
 Keywords Keywords | VIRAL PROTEIN / ASSEMBLIN / UL26 / UL26P / SERINE PROTEASE / PROTEASE / SUID / PRV / HERPES / HERPES VIRUS | |||||||||
| Function / homology |  Function and homology information Function and homology informationassemblin / nuclear capsid assembly / viral release from host cell / host cell cytoplasm / serine-type endopeptidase activity / host cell nucleus / proteolysis / identical protein binding Similarity search - Function | |||||||||
| Biological species |   SUID HERPESVIRUS 1 SUID HERPESVIRUS 1 | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.03 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.03 Å | |||||||||
 Authors Authors | Zuehlsdorf, M. / Werten, S. / Palm, G.J. / Hinrichs, W. | |||||||||
 Citation Citation | Journal: PLoS Pathog / Year: 2015 Title: Dimerization-Induced Allosteric Changes of the Oxyanion-Hole Loop Activate the Pseudorabies Virus Assemblin pUL26N, a Herpesvirus Serine Protease. Authors: Martin Zühlsdorf / Sebastiaan Werten / Barbara G Klupp / Gottfried J Palm / Thomas C Mettenleiter / Winfried Hinrichs /  Abstract: Herpesviruses encode a characteristic serine protease with a unique fold and an active site that comprises the unusual triad Ser-His-His. The protease is essential for viral replication and as such ...Herpesviruses encode a characteristic serine protease with a unique fold and an active site that comprises the unusual triad Ser-His-His. The protease is essential for viral replication and as such constitutes a promising drug target. In solution, a dynamic equilibrium exists between an inactive monomeric and an active dimeric form of the enzyme, which is believed to play a key regulatory role in the orchestration of proteolysis and capsid assembly. Currently available crystal structures of herpesvirus proteases correspond either to the dimeric state or to complexes with peptide mimetics that alter the dimerization interface. In contrast, the structure of the native monomeric state has remained elusive. Here, we present the three-dimensional structures of native monomeric, active dimeric, and diisopropyl fluorophosphate-inhibited dimeric protease derived from pseudorabies virus, an alphaherpesvirus of swine. These structures, solved by X-ray crystallography to respective resolutions of 2.05, 2.10 and 2.03 Å, allow a direct comparison of the main conformational states of the protease. In the dimeric form, a functional oxyanion hole is formed by a loop of 10 amino-acid residues encompassing two consecutive arginine residues (Arg136 and Arg137); both are strictly conserved throughout the herpesviruses. In the monomeric form, the top of the loop is shifted by approximately 11 Å, resulting in a complete disruption of the oxyanion hole and loss of activity. The dimerization-induced allosteric changes described here form the physical basis for the concentration-dependent activation of the protease, which is essential for proper virus replication. Small-angle X-ray scattering experiments confirmed a concentration-dependent equilibrium of monomeric and dimeric protease in solution. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4v08.cif.gz | 187.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4v08.ent.gz | 148.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4v08.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v0/4v08ftp://data.pdbj.org/pub/pdb/validation_reports/v0/4v08 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4cx8C  4v07SC  4v0tC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.09145, 0.02409, 0.9955), Vector: |
-Components
| #1: Protein | Mass: 26645.357 Da / Num. of mol.: 2 / Fragment: RESIDUES 1-224 Source method: isolated from a genetically manipulated source Details: N-TERMINAL (HIS)6-TAG WITH THROMBIN-LINKER / Source: (gene. exp.) SUID HERPESVIRUS 1 / Production host:  #2: Chemical | 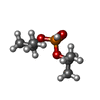  Mass: 166.155 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H15O3P Mass: 166.155 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H15O3P#3: Chemical |   Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl#4: Chemical | ChemComp-MG / |   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 263 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 263 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Sequence details | ASSEMBLIN-PART CRYSTALLIZ | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.07 Å3/Da / Density % sol: 40.63 % Description: NO PHASING WAS NECESSARY SINCE THE CRYSTAL OF THE MODEL AND THE ONE USED FOR THIS DATASET WERE ISOMORPHOUS |
|---|---|
| Crystal grow | pH: 8 Details: PROTEIN SOLUTION WAS INCUBATED WITH 5 MM DFP FOR 1 HOUR, THEN CRYSTALLIZED FROM 0.1 M TRIS/HCL PH 8, 0.2 M MGCL2, 20% PEG 8000 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY / Beamline: 14.1 / Wavelength: 0.91841 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Apr 30, 2014 / Details: MIRRORS |
| Radiation | Monochromator: SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91841 Å / Relative weight: 1 |
| Reflection | Resolution: 2.03→62.8 Å / Num. obs: 29319 / % possible obs: 99.9 % / Observed criterion σ(I): -3 / Redundancy: 6.5 % / Biso Wilson estimate: 35 Å2 / Rmerge(I) obs: 0.11 / Net I/σ(I): 13.08 |
| Reflection shell | Resolution: 2.03→2.14 Å / Redundancy: 6.3 % / Rmerge(I) obs: 0.78 / Mean I/σ(I) obs: 2.2 / % possible all: 99.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4V07 Resolution: 2.03→62.85 Å / Cor.coef. Fo:Fc: 0.965 / Cor.coef. Fo:Fc free: 0.935 / SU B: 9.813 / SU ML: 0.137 / Cross valid method: THROUGHOUT / ESU R: 0.181 / ESU R Free: 0.174 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES WITH TLS ADDED, RESIDUES 1,46,218 OF CHAIN A AND RESIDUES 19,84,114,169,218 OF ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES WITH TLS ADDED, RESIDUES 1,46,218 OF CHAIN A AND RESIDUES 19,84,114,169,218 OF CHAIN B MODELED AS ALA AND RESIDUES 156 AND 169 FROM CHAIN A SHORTENED DUE TO INSUFFICIENT ELECTRON DENSITY, TAG AND THROMBIN-LINKER OF BOTH CHAINS AND RESIDUES 115-119 AND 219-224 OF CHAIN A ARE DISORDERED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 35.46 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.03→62.85 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|