 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4txo: Crystal structure of the mixed disulfide complex of thioredoxin-l... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4txo | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the mixed disulfide complex of thioredoxin-like TlpAs(C110S) and copper chaperone ScoIs(C74S) | ||||||||||||
 Components Components |
| ||||||||||||
 Keywords Keywords | OXIDODREDUCTASE/COPPER BINDING PROTEIN / mixed disulfide / soluble domain of membrane protein / thioredoxin fold / copper protein / protein binding / OXIDODREDUCTASE-COPPER BINDING PROTEIN complex | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationdisulfide oxidoreductase activity / cytochrome complex assembly / thioredoxin peroxidase activity / cellular response to oxidative stress / metal ion binding / plasma membrane Similarity search - Function | ||||||||||||
| Biological species |  Bradyrhizobium diazoefficiens USDA 110 (bacteria) Bradyrhizobium diazoefficiens USDA 110 (bacteria) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.2 Å | ||||||||||||
 Authors Authors | Scharer, M.A. / Abicht, H.K. / Glockshuber, R. / Hennecke, H. | ||||||||||||
| Funding support |  Switzerland, 3items Switzerland, 3items
| ||||||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2014 Title: How Periplasmic Thioredoxin TlpA Reduces Bacterial Copper Chaperone ScoI and Cytochrome Oxidase Subunit II (CoxB) Prior to Metallation. Authors: Abicht, H.K. / Scharer, M.A. / Quade, N. / Ledermann, R. / Mohorko, E. / Capitani, G. / Hennecke, H. / Glockshuber, R. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4txo.cif.gz | 529.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4txo.ent.gz | 433.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4txo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tx/4txoftp://data.pdbj.org/pub/pdb/validation_reports/tx/4txo | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4txvC  1jfuS  1xzoS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 8 molecules CAEGDBFH
| #1: Protein | Mass: 19528.553 Da / Num. of mol.: 4 / Fragment: UNP Residues 38-221 / Mutation: C110S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Bradyrhizobium diazoefficiens USDA 110 (bacteria)Gene: tlpA, bll1380 / Plasmid: pMal-p/TlpASC110S / Production host: #2: Protein | Mass: 19325.930 Da / Num. of mol.: 4 / Fragment: UNP Residues 30-196 / Mutation: C74S, WSHPQFEK: StrepII purification tag Source method: isolated from a genetically manipulated source Source: (gene. exp.) Bradyrhizobium diazoefficiens USDA 110 (bacteria)Gene: blr1131 / Plasmid: pRJ8336 / Production host: |
|---|
-Non-polymers , 4 types, 595 molecules 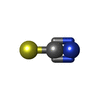



| #3: Chemical |  Mass: 58.082 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CNS Mass: 58.082 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CNS#4: Chemical | ChemComp-PEG / |  Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3#5: Chemical |  Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 590 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 44.87 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 8 Details: Crystals were grown with the sitting-drop vapor diffusion technique by mixing 0.75 ul of the TlpA-ScoI complex (10 mg/ml in 10 mM Tris-HCl, pH 8) with 0.75 ul of precipitant solution (final ...Details: Crystals were grown with the sitting-drop vapor diffusion technique by mixing 0.75 ul of the TlpA-ScoI complex (10 mg/ml in 10 mM Tris-HCl, pH 8) with 0.75 ul of precipitant solution (final concentration: 3% glycerol, 0.2 M NaSCN and 16.0% (w/v) PEG 3350) and equilibration over 100 ul of well solution (3% Glycerol, 0.2 M NaSCN, 28% PEG 3350). |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS / Beamline: X06DA / Wavelength: 0.97941 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 2M / Detector: PIXEL / Date: May 26, 2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Barrtels MONOCHROMATOR WITH DUAL CHANNEL CUT CRYSTALS Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97941 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection twin | Operator: h,-k,-l / Fraction: 0.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.2→50 Å / Num. obs: 68110 / % possible obs: 98.9 % / Observed criterion σ(I): -3 / Redundancy: 7.5 % / Biso Wilson estimate: 40.4 Å2 / Rmerge F obs: 0.996 / Rmerge(I) obs: 0.147 / Rrim(I) all: 0.158 / Χ2: 0.911 / Net I/σ(I): 10.55 / Num. measured all: 512163 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
-Phasing
| Phasing | Method: molecular replacement |
|---|---|
| Phasing MR | Packing: 0 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entries 1jfu, 1xzo Resolution: 2.2→49.338 Å / FOM work R set: 0.8556 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 21.95 / Stereochemistry target values: TWIN_LSQ_F
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 128.77 Å2 / Biso mean: 43.43 Å2 / Biso min: 6.54 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.2→49.338 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 10
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
