 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4qtd: Structure of human JNK1 in complex with SCH772984 and the AMPPNP-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4qtd | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of human JNK1 in complex with SCH772984 and the AMPPNP-hydrolysed triphosphate revealing the second type-I binding mode | ||||||
 Components Components | Mitogen-activated protein kinase 8 | ||||||
 Keywords Keywords | Transferase/transferase inhibitor / Structural Genomics / Structural Genomics Consortium / SGC / TRANSFERASE / kinase / MAPK / signalling / inhibitor / allosteric / Structural Genomics Consortium (SGC) / Transferase-transferase inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of cell killing / JUN phosphorylation / Activation of BMF and translocation to mitochondria / Interleukin-38 signaling / basal dendrite / JUN kinase activity / Activation of BIM and translocation to mitochondria / positive regulation of protein localization to mitochondrion / WNT5:FZD7-mediated leishmania damping / positive regulation of cyclase activity ...positive regulation of cell killing / JUN phosphorylation / Activation of BMF and translocation to mitochondria / Interleukin-38 signaling / basal dendrite / JUN kinase activity / Activation of BIM and translocation to mitochondria / positive regulation of protein localization to mitochondrion / WNT5:FZD7-mediated leishmania damping / positive regulation of cyclase activity / histone deacetylase regulator activity / peptidyl-threonine phosphorylation / NRAGE signals death through JNK / Activation of the AP-1 family of transcription factors / Fc-epsilon receptor signaling pathway / positive regulation of protein metabolic process / positive regulation of NLRP3 inflammasome complex assembly / mitogen-activated protein kinase / regulation of macroautophagy / negative regulation of protein binding / response to mechanical stimulus / stress-activated MAPK cascade / response to UV / JNK cascade / energy homeostasis / cellular response to amino acid starvation / NRIF signals cell death from the nucleus / peptidyl-serine phosphorylation / negative regulation of autophagy / integrin-mediated signaling pathway / JNK (c-Jun kinases) phosphorylation and activation mediated by activated human TAK1 / protein serine/threonine kinase binding / FCERI mediated MAPK activation / cellular response to reactive oxygen species / cellular response to mechanical stimulus / regulation of circadian rhythm / autophagy / histone deacetylase binding / cellular senescence / Signaling by ALK fusions and activated point mutants / rhythmic process / MAPK cascade / regulation of protein localization / Recruitment and ATM-mediated phosphorylation of repair and signaling proteins at DNA double strand breaks / cellular response to lipopolysaccharide / cellular response to oxidative stress / response to oxidative stress / protein phosphatase binding / Oxidative Stress Induced Senescence / protein phosphorylation / positive regulation of apoptotic process / protein serine kinase activity / axon / protein serine/threonine kinase activity / positive regulation of gene expression / synapse / negative regulation of apoptotic process / enzyme binding / nucleoplasm / ATP binding / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | ||||||
 Authors Authors | Chaikuad, A. / Keates, T. / von Delft, F. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Knapp, S. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Nat.Chem.Biol. / Year: 2014 Title: A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Authors: Chaikuad, A. / M C Tacconi, E. / Zimmer, J. / Liang, Y. / Gray, N.S. / Tarsounas, M. / Knapp, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4qtd.cif.gz | 176.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4qtd.ent.gz | 137.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4qtd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qt/4qtdftp://data.pdbj.org/pub/pdb/validation_reports/qt/4qtd | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4qtaC  4qtbC  4qtcC  4qteC  2ydiS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |


















-Links
 PDBj
PDBj















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 42054.707 Da / Num. of mol.: 1 / Fragment: kinase domain (1-363) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: JNK1, MAPK8, PRKM8, SAPK1, SAPK1C / Plasmid: pNIC28-Bsa4 / Production host:  References: UniProt: P45983, mitogen-activated protein kinase |
|---|
-Non-polymers , 6 types, 393 molecules 

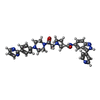



| #2: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 22 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 22 / Source method: obtained synthetically / Formula: C2H6O2#3: Chemical | ChemComp-EPE / |  Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM#4: Chemical | ChemComp-38Z / ( |  Mass: 587.674 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H33N9O2 Mass: 587.674 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H33N9O2#5: Chemical | ChemComp-ANP / |  Mass: 506.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O12P3 / Comment: AMP-PNP, energy-carrying molecule analogue*YM Mass: 506.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O12P3 / Comment: AMP-PNP, energy-carrying molecule analogue*YM#6: Chemical | ChemComp-MG / |  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 367 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.34 Å3/Da / Density % sol: 47.53 % |
|---|---|
| Crystal grow | Temperature: 277.15 K / Method: vapor diffusion, sitting drop Details: 12-15% PEG3350 and 0.1 M HEPES pH 6.8-7.8, VAPOR DIFFUSION, SITTING DROP, temperature 277.15K PH range: 6.8-7.8 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 0.97625 Å / Beamline: I04 / Wavelength: 0.97625 Å |
| Detector | Type: PSI PILATUS 6M / Detector: PIXEL / Date: Apr 14, 2014 / Details: Kirkpatrick Baez bimorph mirror pair |
| Radiation | Monochromator: Si (111) double crystal monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97625 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→25.75 Å / Num. all: 64041 / Num. obs: 63969 / % possible obs: 99.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 6.4 % / Biso Wilson estimate: 15.2 Å2 / Rmerge(I) obs: 0.064 / Net I/σ(I): 14.9 |
| Reflection shell | Resolution: 1.5→1.58 Å / Redundancy: 4.9 % / Rmerge(I) obs: 0.497 / Mean I/σ(I) obs: 3.1 / Num. unique all: 9131 / % possible all: 99.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entry 2YDI Resolution: 1.5→59.68 Å / Cor.coef. Fo:Fc: 0.97 / Cor.coef. Fo:Fc free: 0.959 / SU B: 2.639 / SU ML: 0.049 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 2 / ESU R: 0.069 / ESU R Free: 0.071 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.467 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.171 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→59.68 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.5→1.539 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
