- PDB-4qm8: Crystal Structure of a Putative Cysteine Dioxygnase From Bacillus... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4qm8
Title
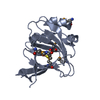
Crystal Structure of a Putative Cysteine Dioxygnase From Bacillus subtilis: A Alternative Modeling of 3EQE
Components
Cysteine dioxygenase
Keywords
OXIDOREDUCTASE / Cupin Fold / Cysteine dioxygnease
Function / homology
Function and homology information
cysteine dioxygenase / cysteine dioxygenase activity / oxidoreductase activity, acting on single donors with incorporation of molecular oxygen, incorporation of two atoms of oxygen / ferrous iron binding Similarity search - Function
Cysteine dioxygenase type I / Cysteine dioxygenase type I / RmlC-like cupin domain superfamily / Jelly Rolls / RmlC-like jelly roll fold / Jelly Rolls / Sandwich / Mainly Beta Similarity search - Domain/homology
THIS ENTRY 4QM8 REFLECTS AN ALTERNATIVE MODELING OF THE ORIGINAL STRUCTURAL DATA R3EQESF DETERMINED ...THIS ENTRY 4QM8 REFLECTS AN ALTERNATIVE MODELING OF THE ORIGINAL STRUCTURAL DATA R3EQESF DETERMINED BY AUTHORS OF THE PDB ENTRY 3EQE:S.M.VOROBIEV,M.SU,J.SEETHARAMAN,J.BENACH,F.FOROUHAR, G.CLAYTON,B.COOPER,H.WANG,E.L.FOOTE,C.CICCOSANTI,L.MAO, R.XIAO,T.B.ACTON,G.T.MONTELIONE,J.F.HUNT,L.TONG,NORTHEAST STRUCTURAL GENOMICS CONSORTIUM (NESG)
Mass: 18.015 Da / Num. of mol.: 63 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
Nonpolymer details
AUTHORS HAVE DECLARED THAT THE ACTIVE SITE DENSITY AT THIS RESOLUTION IS NOT DEFINITIVE, SO TO BE ...AUTHORS HAVE DECLARED THAT THE ACTIVE SITE DENSITY AT THIS RESOLUTION IS NOT DEFINITIVE, SO TO BE CONSERVATIVE OUR FINAL REFINEMENT AND DEPOSITION USED THE MINIMAL INTERPRETATION OF THREE WATERS IN THE ACTIVE SITE. TO ENSURE THAT USERS OF THE COORDINATES ARE AWARE THAT CYS MAY BE BOUND, WE ALSO INCLUDE IN THE FILE, WITH OCCUPANCY SET TO ZERO, THE COORDINATES AND B-FACTORS WE OBTAINED BY REFINING A BOUND CYS AT 100% OCCUPANCY
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.68 Å3/Da / Density % sol: 54.18 % / Description: AUTHOR USED THE SF DATA FROM ENTRY 3EQE.
-
Data collection
Radiation
Scattering type: x-ray
Radiation wavelength
Relative weight: 1
-
Processing
Software
Name
Version
Classification
SHELXDE
phasing
PHENIX
(phenix.refine: 1.9_1692)
refinement
Refinement
Resolution: 2.82→46.51 Å / SU ML: 0.39 / σ(F): 1.98 / Phase error: 26.94 / Stereochemistry target values: ML
Rfactor
Num. reflection
% reflection
Rfree
0.254
465
4.19 %
Rwork
0.177
-
-
obs
0.18
11097
99.6 %
all
-
19930
-
Solvent computation
Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL
Refinement step
Cycle: LAST / Resolution: 2.82→46.51 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
2352
0
16
63
2431
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.011
2416
X-RAY DIFFRACTION
f_angle_d
1.174
3273
X-RAY DIFFRACTION
f_dihedral_angle_d
14.395
891
X-RAY DIFFRACTION
f_chiral_restr
0.042
371
X-RAY DIFFRACTION
f_plane_restr
0.005
427
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
2.8203-3.2283
0.303
147
0.2205
3408
X-RAY DIFFRACTION
99
3.2283-4.0669
0.277
152
0.1982
3498
X-RAY DIFFRACTION
100
4.0669-46.5197
0.2318
166
0.1562
3726
X-RAY DIFFRACTION
100
Refinement TLS params.
Method: refined / Refine-ID: X-RAY DIFFRACTION
ID
L11 (°2)
L12 (°2)
L13 (°2)
L22 (°2)
L23 (°2)
L33 (°2)
S11 (Å °)
S12 (Å °)
S13 (Å °)
S21 (Å °)
S22 (Å °)
S23 (Å °)
S31 (Å °)
S32 (Å °)
S33 (Å °)
T11 (Å2)
T12 (Å2)
T13 (Å2)
T22 (Å2)
T23 (Å2)
T33 (Å2)
Origin x (Å)
Origin y (Å)
Origin z (Å)
1
8.2144
3.0178
-0.4006
7.1499
-0.194
3.7545
0.0274
-0.0136
0.1388
-0.0907
0.0361
-0.4668
0.0037
0.168
-0.0267
0.5886
0.0152
0.0416
0.3245
-0.0065
0.3469
-39.7633
-19.2955
141.5321
2
7.0365
0.9998
-0.3336
3.9853
-1.8359
3.934
0.2318
0.1474
-0.4566
-0.0828
-0.1207
0.0459
0.5831
0.005
-0.1291
0.688
-0.034
0.0515
0.372
-0.0735
0.4085
-39.1092
1.9788
121.6725
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection details
1
X-RAY DIFFRACTION
1
chainA
2
X-RAY DIFFRACTION
2
chainB
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION / Resolution: 2.82 Å
X-RAY DIFFRACTION / Resolution: 2.82 Å  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads














 PDBj
PDBj

 Assembly
Assembly


 Mass: 55.845 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe
Mass: 55.845 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe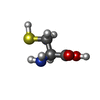
 Type: L-peptide linking / Mass: 121.158 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H7NO2S
Type: L-peptide linking / Mass: 121.158 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H7NO2S Mass: 18.015 Da / Num. of mol.: 63 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 63 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing