 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4qjh: Crystal Structure of the Twister Ribozyme with the Nucleotide 5'-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4qjh | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Twister Ribozyme with the Nucleotide 5'- to the Cleavage Site Ordered at 4.1 A Resolution | ||||||
 Components Components | (Twister Ribozyme) x 2 | ||||||
 Keywords Keywords | RNA / small self-cleaving ribozyme | ||||||
| Function / homology | RNA / RNA (> 10) Function and homology information Function and homology information | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MIRAS / Resolution: 3.88 Å X-RAY DIFFRACTION / SYNCHROTRON / MIRAS / Resolution: 3.88 Å | ||||||
 Authors Authors | Eiler, D.R. / Wang, J. / Steitz, T.A. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2014 Title: Structural basis for the fast self-cleavage reaction catalyzed by the twister ribozyme. Authors: Eiler, D. / Wang, J. / Steitz, T.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4qjh.cif.gz | 131 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4qjh.ent.gz | 101.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4qjh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qj/4qjhftp://data.pdbj.org/pub/pdb/validation_reports/qj/4qjh | HTTPS FTP |
|---|
-Related structure data









-Links
 PDBj
PDBj





























- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| 2 | 
| |||||||||
| 3 | 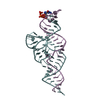
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: RNA chain | Mass: 8257.906 Da / Num. of mol.: 3 / Source method: obtained synthetically #2: RNA chain | Mass: 15723.355 Da / Num. of mol.: 3 / Source method: obtained synthetically Details: RNA was prepared by in vitro transcription with T7 RNA polymerase. #3: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 3 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5.31 Å3/Da / Density % sol: 76.82 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 4.6 Details: 160 mM sodium citrate, pH 4.6, 700 mM ammonium sulfate, 1 M lithium sulfate, 3% pentaerythriotol ethoxylate (3/4 EO/OH), 3% 6-aminohexanoic acid, VAPOR DIFFUSION, SITTING DROP, temperature 298K |
-Data collection
| Diffraction |
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||||||||||||||||||||||||||
| Detector |
| ||||||||||||||||||||||||||||||||||||||||||
| Radiation |
| ||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.018→198.121 Å / Num. all: 99843 / Num. obs: 12154 / % possible obs: 99.9 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 20.6 % / Rmerge(I) obs: 0.071 / Net I/σ(I): 19 | ||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|

- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIRAS / Resolution: 3.88→198.12 Å / Cor.coef. Fo:Fc: 0.968 / Cor.coef. Fo:Fc free: 0.943 / SU B: 40.977 / SU ML: 0.512 / Cross valid method: THROUGHOUT / ESU R Free: 0.53 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 146.821 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.88→198.12 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|