 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4nvc: Predicting protein conformational response in prospective ligand ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4nvc | ||||||
|---|---|---|---|---|---|---|---|
| Title | Predicting protein conformational response in prospective ligand discovery | ||||||
 Components Components | Cytochrome c peroxidase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / Model system / flexibility / dynamic / loop / side-chains / energy penalty / occupancy / Boltzmann weights / flexible docking / ligand binding | ||||||
| Function / homology |  Function and homology information Function and homology informationcytochrome-c peroxidase activity / Oxidoreductases; Acting on a peroxide as acceptor; Peroxidases / response to reactive oxygen species / hydrogen peroxide catabolic process / mitochondrial intermembrane space / cellular response to oxidative stress / mitochondrial matrix / heme binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.6 Å | ||||||
 Authors Authors | Fischer, M. / Fraser, J.S. | ||||||
 Citation Citation | Journal: Nat Chem / Year: 2014 Title: Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Authors: Fischer, M. / Coleman, R.G. / Fraser, J.S. / Shoichet, B.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4nvc.cif.gz | 145.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4nvc.ent.gz | 113.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4nvc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nv/4nvcftp://data.pdbj.org/pub/pdb/validation_reports/nv/4nvc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4nvaC  4nvbC  4nvdC  4nveC  4nvfC  4nvgC  4nvhC  4nviC  4nvjC  4nvkC  4nvlC  4nvmC  4nvnC  4nvoC  4oq7C C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 32928.582 Da / Num. of mol.: 1 / Fragment: UNP RESIDUES 72-362 / Mutation: P190G, W191G, DELETIONS G192-A193 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: RM11-1a / Gene: CCP1 CCP CPO YKR066C, SCRG_04081 / Plasmid details: 469008 / Production host:  |
|---|---|
| #2: Chemical | ChemComp-BEN / 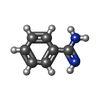  Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 |
| #3: Chemical | ChemComp-HEM /   Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 383 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 383 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.98 Å3/Da / Density % sol: 58.71 % |
|---|---|
| Crystal grow | Temperature: 283 K / Method: vapor diffusion, hanging drop / pH: 6 Details: Compound soaked into crystal grown in equal volume of 500mM MES buffer (pH 6.0) and 25% MPD, VAPOR DIFFUSION, HANGING DROP, temperature 283K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.3.1 / Wavelength: 1.11587 Å / Beamline: 8.3.1 / Wavelength: 1.11587 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Oct 25, 2011 |
| Radiation | Monochromator: KOHZU DUAL DOUBLE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.11587 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→31.5 Å / Num. all: 44139 / Num. obs: 44139 / % possible obs: 84.8 % / Observed criterion σ(F): 1.35 / Observed criterion σ(I): 2.1 / Redundancy: 3.6 % / Biso Wilson estimate: 14.6 Å2 / Rmerge(I) obs: 0.04 / Net I/σ(I): 20 |
| Reflection shell | Resolution: 1.6→1.65 Å / Redundancy: 2.1 % / Rmerge(I) obs: 0.1 / Mean I/σ(I) obs: 6.3 / Num. unique all: 1630 / % possible all: 84.8 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.6→31.5 Å / Occupancy max: 1 / Occupancy min: 0.12 / FOM work R set: 0.9037 / SU ML: 0.13 / σ(F): 1.35 / Phase error: 16.85 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 65.07 Å2 / Biso mean: 17.6804 Å2 / Biso min: 5.98 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→31.5 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 16
|
