Entry Database : PDB / ID : 4nahTitle Inhibitors of 4-Phosphopanthetheine Adenylyltransferase (PPAT) Phosphopantetheine adenylyltransferase Keywords / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / Biological species Staphylococcus aureus (bacteria)Method / / / Resolution : 2.38 Å Authors Lahiri, S.D. Journal : Antimicrob.Agents Chemother. / Year : 2013Title : Discovery of inhibitors of 4'-phosphopantetheine adenylyltransferase (PPAT) to validate PPAT as a target for antibacterial therapy.Authors: de Jonge, B.L. / Walkup, G.K. / Lahiri, S.D. / Huynh, H. / Neckermann, G. / Utley, L. / Nash, T.J. / Brock, J. / San Martin, M. / Kutschke, A. / Johnstone, M. / Laganas, V. / Hajec, L. / Gu, ... Authors : de Jonge, B.L. / Walkup, G.K. / Lahiri, S.D. / Huynh, H. / Neckermann, G. / Utley, L. / Nash, T.J. / Brock, J. / San Martin, M. / Kutschke, A. / Johnstone, M. / Laganas, V. / Hajec, L. / Gu, R.F. / Ni, H. / Chen, B. / Hutchings, K. / Holt, E. / McKinney, D. / Gao, N. / Livchak, S. / Thresher, J. History Deposition Oct 22, 2013 Deposition site / Processing site Revision 1.0 Mar 12, 2014 Provider / Type Revision 1.1 Feb 28, 2024 Group / Database references / Derived calculationsCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Staphylococcus aureus (bacteria)
Staphylococcus aureus (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj
 Assembly
Assembly

 Mass: 523.247 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H16N5O12P3S / Comment: ATP-gamma-S, energy-carrying molecule analogue*YM
Mass: 523.247 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H16N5O12P3S / Comment: ATP-gamma-S, energy-carrying molecule analogue*YM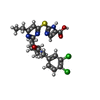
 Mass: 534.458 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C24H25Cl2N5O3S
Mass: 534.458 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C24H25Cl2N5O3S Mass: 18.015 Da / Num. of mol.: 78 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 78 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 19-BM / Wavelength: 1.0723
/ Beamline: 19-BM / Wavelength: 1.0723  Processing
Processing