 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4mh7: Crystal structure of the catalytic domain of the proto-oncogene t... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4mh7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the catalytic domain of the proto-oncogene tyrosine-protein kinase MER in complex with inhibitor UNC1896 | ||||||
 Components Components | Tyrosine-protein kinase Mer | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / TYROSINE KINASE / ACUTE LYMPHOBLASTIC LEUKEMIA / RATIONAL STRUCTURE BASED DRUG DESIGN / TRANSFERASE-TRANSFERASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of leukocyte apoptotic process / Dengue Virus Attachment and Entry / photoreceptor outer segment / phagocytosis / transmembrane receptor protein tyrosine kinase activity / positive regulation of phagocytosis / cell surface receptor protein tyrosine kinase signaling pathway / Cell surface interactions at the vascular wall / receptor protein-tyrosine kinase / cell-cell signaling ...negative regulation of leukocyte apoptotic process / Dengue Virus Attachment and Entry / photoreceptor outer segment / phagocytosis / transmembrane receptor protein tyrosine kinase activity / positive regulation of phagocytosis / cell surface receptor protein tyrosine kinase signaling pathway / Cell surface interactions at the vascular wall / receptor protein-tyrosine kinase / cell-cell signaling / cell migration / nervous system development / protein phosphorylation / signaling receptor complex / cell surface receptor signaling pathway / : / ATP binding / plasma membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.51 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.51 Å | ||||||
 Authors Authors | Zhang, W. / McIver, A. / Stashko, M.A. / Deryckere, D. / Branchford, B.R. / Hunter, D. / Kireev, D.B. / Miley, M.J. / Norris-Drouin, J. / Stewart, W.M. ...Zhang, W. / McIver, A. / Stashko, M.A. / Deryckere, D. / Branchford, B.R. / Hunter, D. / Kireev, D.B. / Miley, M.J. / Norris-Drouin, J. / Stewart, W.M. / Lee, M. / Sather, S. / Zhou, Y. / DiPaola, J.A. / Machius, M. / Janzen, W.P. / Earp, H.S. / Graham, D.K. / Frye, S. / Wang, X. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2013 Title: Discovery of Mer specific tyrosine kinase inhibitors for the treatment and prevention of thrombosis. Authors: Zhang, W. / McIver, A.L. / Stashko, M.A. / DeRyckere, D. / Branchford, B.R. / Hunter, D. / Kireev, D. / Miley, M.J. / Norris-Drouin, J. / Stewart, W.M. / Lee, M. / Sather, S. / Zhou, Y. / Di ...Authors: Zhang, W. / McIver, A.L. / Stashko, M.A. / DeRyckere, D. / Branchford, B.R. / Hunter, D. / Kireev, D. / Miley, M.J. / Norris-Drouin, J. / Stewart, W.M. / Lee, M. / Sather, S. / Zhou, Y. / Di Paola, J.A. / Machius, M. / Janzen, W.P. / Earp, H.S. / Graham, D.K. / Frye, S.V. / Wang, X. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4mh7.cif.gz | 201.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4mh7.ent.gz | 161.9 KB | Display | PDB format |
| PDBx/mmJSON format | 4mh7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mh/4mh7ftp://data.pdbj.org/pub/pdb/validation_reports/mh/4mh7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4mhaC  3brbS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |

















-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 35889.434 Da / Num. of mol.: 2 / Fragment: CATALYTIC DOMAIN, UNP RESIDUES 570-864 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: MER, MERTK / Plasmid: PET28 / Production host:  References: UniProt: Q12866, receptor protein-tyrosine kinase #2: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Cl#3: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#4: Chemical | ChemComp-MH7 / | 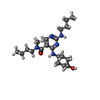  Mass: 377.524 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H35N5O2 Mass: 377.524 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H35N5O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 29 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 29 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.21 Å3/Da / Density % sol: 44.26 % |
|---|---|
| Crystal grow | Temperature: 288 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: Protein: 32.5 mg/mL in 20 mM Tris pH 8.0, 500mM sodium chloride, 2mM beta-mercaptoethanol, incubated with inhibitor (2.5 mM final concentration) overnight, mixed 1:1 with crystallization ...Details: Protein: 32.5 mg/mL in 20 mM Tris pH 8.0, 500mM sodium chloride, 2mM beta-mercaptoethanol, incubated with inhibitor (2.5 mM final concentration) overnight, mixed 1:1 with crystallization solution (27-33% (v/v) Peg400, 200 mM magnesium chloride, 100 mM Tris pH 8.5), VAPOR DIFFUSION, SITTING DROP, temperature 288K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-ID / Wavelength: 0.97921 Å / Beamline: 22-ID / Wavelength: 0.97921 Å |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Jun 13, 2012 |
| Radiation | Monochromator: SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97921 Å / Relative weight: 1 |
| Reflection | Resolution: 2.51→32.06 Å / Num. all: 20799 / Num. obs: 20799 / % possible obs: 97.7 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 4.1 % / Biso Wilson estimate: 31.2 Å2 / Rmerge(I) obs: 0.125 / Net I/σ(I): 10.7 |
| Reflection shell | Resolution: 2.51→2.53 Å / Redundancy: 3.7 % / Rmerge(I) obs: 0.564 / Mean I/σ(I) obs: 2.2 / Num. unique all: 428 / % possible all: 81.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 3BRB Resolution: 2.51→32.06 Å / Cross valid method: Maximum Likelihood / σ(F): 0 / Stereochemistry target values: ML
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.51→32.06 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.51→2.58 Å
|
