 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4feu: Crystal structure of the aminoglycoside phosphotransferase APH(3'... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4feu | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the aminoglycoside phosphotransferase APH(3')-Ia, with substrate kanamycin and small molecule inhibitor anthrapyrazolone SP600125 | ||||||
 Components Components | Aminoglycoside 3'-phosphotransferase AphA1-IAB | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / anthrapyrazolone / SP600125 / protein kinase inhibitor / CENTER FOR STRUCTURAL GENOMICS OF INFECTIOUS DISEASES / CSGID / NIAID / NATIONAL INSTITUTE OF ALLERGY AND INFECTIOUS DISEASES / EUKARYOTIC PROTEIN KINASE-LIKE FOLD / ALPHA/BETA PROTEIN / TRANSFERASE / PHOSPHOTRANSFERASE / AMINOGLYCOSIDE PHOSPHOTRANSFERASE / ANTIBIOTIC RESISTANCE / AMINOGLYCOSIDES / KANAMYCIN / GTP / INTRACELLULAR / TRANSFERASE-TRANSFERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationkanamycin kinase / kanamycin kinase activity / phosphorylation / response to antibiotic / ATP binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  Acinetobacter baumannii (bacteria) Acinetobacter baumannii (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.37 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.37 Å | ||||||
 Authors Authors | Stogios, P.J. / Evdokimova, E. / Wawrzak, Z. / Minasov, G. / Egorova, O. / Di Leo, R. / Shakya, T. / Spanogiannopoulos, P. / Wright, G.D. / Savchenko, A. ...Stogios, P.J. / Evdokimova, E. / Wawrzak, Z. / Minasov, G. / Egorova, O. / Di Leo, R. / Shakya, T. / Spanogiannopoulos, P. / Wright, G.D. / Savchenko, A. / Anderson, W.F. / Center for Structural Genomics of Infectious Diseases (CSGID) | ||||||
 Citation Citation | Journal: Biochem.J. / Year: 2013 Title: Structure-guided optimization of protein kinase inhibitors reverses aminoglycoside antibiotic resistance. Authors: Stogios, P.J. / Spanogiannopoulos, P. / Evdokimova, E. / Egorova, O. / Shakya, T. / Todorovic, N. / Capretta, A. / Wright, G.D. / Savchenko, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4feu.cif.gz | 648.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4feu.ent.gz | 540.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4feu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fe/4feuftp://data.pdbj.org/pub/pdb/validation_reports/fe/4feu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4ej7SC  4fevC  4fewC  4fexC  4gkhC  4gkiC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| 5 | 
| ||||||||
| 6 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 31350.404 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Acinetobacter baumannii (bacteria) / Strain: AYE / Gene: ABAYE3578, APHA1-IAB / Plasmid: P15TV LIC / Production host: #2: Chemical | ChemComp-KAN /   Mass: 484.499 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C18H36N4O11 / Comment: antibiotic*YM Mass: 484.499 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C18H36N4O11 / Comment: antibiotic*YM#3: Chemical | 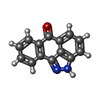  Mass: 220.226 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C14H8N2O Mass: 220.226 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C14H8N2O#4: Chemical | ChemComp-ACT /   Mass: 59.044 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H3O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 675 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 675 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.36 Å3/Da / Density % sol: 47.79 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 4.5 Details: 0.1 M sodium acetate pH 4.5, 8% PEG3350, 3% DMSO, 0.2 M NDSB221, 2 mM kanamycin, 3 mM SP600125, VAPOR DIFFUSION, SITTING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 21-ID-G / Wavelength: 0.97856 Å / Beamline: 21-ID-G / Wavelength: 0.97856 Å |
| Detector | Type: MAR scanner 300 mm plate / Detector: IMAGE PLATE / Date: Mar 21, 2011 / Details: BERYLLIUM LENSES |
| Radiation | Monochromator: DIAMOND / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97856 Å / Relative weight: 1 |
| Reflection | Resolution: 2.37→20 Å / Num. obs: 67481 / % possible obs: 98.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -2 / Redundancy: 2.7 % / Biso Wilson estimate: 50.58 Å2 / Rsym value: 0.072 / Net I/σ(I): 16.35 |
| Reflection shell | Resolution: 2.37→2.41 Å / Redundancy: 2.7 % / Mean I/σ(I) obs: 2 / Rsym value: 0.423 / % possible all: 98.4 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB 4EJ7 Resolution: 2.37→19.97 Å / Cor.coef. Fo:Fc: 0.954 / Cor.coef. Fo:Fc free: 0.9257 / Cross valid method: THROUGHOUT / σ(F): 0
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 55.88 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.279 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.37→19.97 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.37→2.43 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
