 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4ekv: Streptavidin 8-aa-loop H127C mutein with reversible biotin binding -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4ekv | ||||||
|---|---|---|---|---|---|---|---|
| Title | Streptavidin 8-aa-loop H127C mutein with reversible biotin binding | ||||||
 Components Components | Streptavidin | ||||||
 Keywords Keywords | biotin-binding protein / Beta-barrel / Binding protein / Biotin-binding | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Streptomyces avidinii (bacteria) Streptomyces avidinii (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2 Å | ||||||
 Authors Authors | Barrette-Ng, I.H. / Honetschlaeger, C. / Wong, S.L. / Ng, K.K.S. | ||||||
 Citation Citation | Journal: Plos One / Year: 2012 Title: Development of a tetrameric streptavidin mutein with reversible biotin binding capability: engineering a mobile loop as an exit door for biotin. Authors: O'Sullivan, V.J. / Barrette-Ng, I. / Hommema, E. / Hermanson, G.T. / Schofield, M. / Wu, S.C. / Honetschlaeger, C. / Ng, K.K. / Wong, S.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4ekv.cif.gz | 39 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4ekv.ent.gz | 25.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4ekv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ek/4ekvftp://data.pdbj.org/pub/pdb/validation_reports/ek/4ekv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1sweS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
| ||||||||
| Details | The homotetramer is generated from the single protomer in the asymmetric unit using the four-fold crystallographic symmetry operators. |
-Components
| #1: Protein | Mass: 16286.493 Da / Num. of mol.: 1 / Mutation: H151C Source method: isolated from a genetically manipulated source Details: Synthetic gene / Source: (gene. exp.) Streptomyces avidinii (bacteria) / Plasmid: pSSAV / Production host: |
|---|---|
| #2: Chemical | ChemComp-BTN / 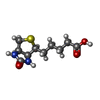  Mass: 244.311 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N2O3S Mass: 244.311 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N2O3S |
| #3: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 70 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 70 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Sequence details | the 8-residue loop region was modified from TTEANAWK to DSSNGSDG |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.75 Å3/Da / Density % sol: 29.88 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6 Details: 50% Tacsimate, 10% glycerol, pH 6.0, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 Å |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Dec 7, 2010 / Details: Osmic multilayer |
| Radiation | Monochromator: Osmic multilayer / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.95→20 Å / Num. all: 8958 / Num. obs: 8607 / % possible obs: 96.1 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 6.7 % / Biso Wilson estimate: 35.1 Å2 / Rmerge(I) obs: 0.063 / Rsym value: 0.063 / Net I/σ(I): 28.4 |
| Reflection shell | Resolution: 1.95→2.02 Å / Redundancy: 3.6 % / Rmerge(I) obs: 0.496 / Mean I/σ(I) obs: 2.8 / Num. unique all: 776 / Rsym value: 0.0496 / % possible all: 88 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entry 1SWE Resolution: 2→19.39 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.939 / WRfactor Rfree: 0.2252 / WRfactor Rwork: 0.1882 / Occupancy max: 1 / Occupancy min: 0.25 / FOM work R set: 0.8437 / SU B: 4.098 / SU ML: 0.114 / SU R Cruickshank DPI: 0.1995 / SU Rfree: 0.1652 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.199 / ESU R Free: 0.165 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: U VALUES: REFINED INDIVIDUALLY. THE SF DATA DO NOT HAVE FREE SET MARKER.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 83.47 Å2 / Biso mean: 37.5624 Å2 / Biso min: 21.08 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→19.39 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.052 Å / Total num. of bins used: 20
|
