 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1swe | ||||||
|---|---|---|---|---|---|---|---|
| Title | APO-CORE-STREPTAVIDIN IN COMPLEX WITH BIOTIN AT PH 4.5 | ||||||
 Components Components | STREPTAVIDIN | ||||||
 Keywords Keywords | BIOTIN-BINDING PROTEIN / BIOTIN BINDING PROTEIN / BIOTIN | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Streptomyces avidinii (bacteria) Streptomyces avidinii (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.06 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.06 Å | ||||||
 Authors Authors | Freitag, S. / Le Trong, I. / Klumb, L. / Stayton, P.S. / Stenkamp, R.E. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 1997 Title: Structural studies of the streptavidin binding loop. Authors: Freitag, S. / Le Trong, I. / Klumb, L. / Stayton, P.S. / Stenkamp, R.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1swe.cif.gz | 106.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1swe.ent.gz | 82.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1swe.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sw/1sweftp://data.pdbj.org/pub/pdb/validation_reports/sw/1swe | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1swaSC  1swbC  1swcC  1swdC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 13281.336 Da / Num. of mol.: 4 / Fragment: CORE, RESIDUES 13 - 139 Source method: isolated from a genetically manipulated source Details: PH 4.5, COMPLEXED WITH BIOTIN / Source: (gene. exp.) Streptomyces avidinii (bacteria) / Production host: #2: Chemical | ChemComp-BTN / 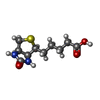  Mass: 244.311 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H16N2O3S Mass: 244.311 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H16N2O3S#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 302 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 302 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.39 Å3/Da / Density % sol: 48.48 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 4.5 Details: PROTEIN-BIOTIN COMPLEX WAS CO-CRYSTALLIZED FROM 50% MPD (2-METHYL-PENTANE-2,4-DIOLE, PH 4.5) WITH 2.5M EXCESS OF BIOTIN | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 295 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: Feb 13, 1996 |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→50 Å / Num. obs: 24738 / % possible obs: 83 % / Redundancy: 11 % / Rmerge(I) obs: 0.066 / Net I/σ(I): 9 |
| Reflection shell | Resolution: 2.1→2.2 Å / Rmerge(I) obs: 0.192 / Mean I/σ(I) obs: 3.4 / % possible all: 42 |
| Reflection shell | *PLUS % possible obs: 42 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1SWA Resolution: 2.06→8 Å / Num. parameters: 15579 / Num. restraintsaints: 15139 / Stereochemistry target values: ENGH AND HUBER
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201 | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 0 / Occupancy sum hydrogen: 3355 / Occupancy sum non hydrogen: 3891 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.06→8 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-96 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rwork: 0.161 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: s_plane_restr / Dev ideal: 0.017 |
