 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4drh: Co-crystal structure of the PPIase domain of FKBP51, Rapamycin an... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4drh | ||||||
|---|---|---|---|---|---|---|---|
| Title | Co-crystal structure of the PPIase domain of FKBP51, Rapamycin and the FRB fragment of mTOR at low pH | ||||||
 Components Components |
| ||||||
 Keywords Keywords | Isomerase/Transferase / Fk-506 binding domain / Hsp90 cochaperone / immunophilin / peptidyl-prolyl isomerase / mammalian target of Rapamycin / kinase / signalling / immunosuppression / cancer / Isomerase-Transferase complex | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of SCF-dependent proteasomal ubiquitin-dependent catabolic process / RNA polymerase III type 2 promoter sequence-specific DNA binding / RNA polymerase III type 1 promoter sequence-specific DNA binding / positive regulation of cytoplasmic translational initiation / regulation of locomotor rhythm / T-helper 1 cell lineage commitment / positive regulation of pentose-phosphate shunt / positive regulation of wound healing, spreading of epidermal cells / regulation of membrane permeability / TORC2 complex ...positive regulation of SCF-dependent proteasomal ubiquitin-dependent catabolic process / RNA polymerase III type 2 promoter sequence-specific DNA binding / RNA polymerase III type 1 promoter sequence-specific DNA binding / positive regulation of cytoplasmic translational initiation / regulation of locomotor rhythm / T-helper 1 cell lineage commitment / positive regulation of pentose-phosphate shunt / positive regulation of wound healing, spreading of epidermal cells / regulation of membrane permeability / TORC2 complex / cellular response to leucine starvation / TFIIIC-class transcription factor complex binding / heart valve morphogenesis / voluntary musculoskeletal movement / negative regulation of lysosome organization / TORC1 complex / calcineurin-NFAT signaling cascade / RNA polymerase III type 3 promoter sequence-specific DNA binding / positive regulation of transcription of nucleolar large rRNA by RNA polymerase I / positive regulation of keratinocyte migration / regulation of osteoclast differentiation / energy reserve metabolic process / MTOR signalling / regulation of lysosome organization / cellular response to L-leucine / Energy dependent regulation of mTOR by LKB1-AMPK / cellular response to nutrient / regulation of autophagosome assembly / Amino acids regulate mTORC1 / cellular response to methionine / negative regulation of cell size / TORC2 signaling / Modulation of host responses by IFN-stimulated genes / cellular response to osmotic stress / cell projection organization / anoikis / inositol hexakisphosphate binding / response to alcohol / cardiac muscle cell development / negative regulation of calcineurin-NFAT signaling cascade / positive regulation of ubiquitin-dependent protein catabolic process / regulation of myelination / negative regulation of protein localization to nucleus / positive regulation of transcription by RNA polymerase III / positive regulation of ruffle assembly / regulation of cell size / FK506 binding / positive regulation of myotube differentiation / negative regulation of macroautophagy / Macroautophagy / Constitutive Signaling by AKT1 E17K in Cancer / positive regulation of actin filament polymerization / germ cell development / oligodendrocyte differentiation / TORC1 signaling / positive regulation of oligodendrocyte differentiation / behavioral response to pain / response to amino acid / TOR signaling / mTORC1-mediated signalling / CD28 dependent PI3K/Akt signaling / HSF1-dependent transactivation / regulation of macroautophagy / positive regulation of translational initiation / positive regulation of epithelial to mesenchymal transition / positive regulation of lipid biosynthetic process / 'de novo' pyrimidine nucleobase biosynthetic process / vascular endothelial cell response to laminar fluid shear stress / heart morphogenesis / regulation of cellular response to heat / positive regulation of lamellipodium assembly / neuronal action potential / T cell costimulation / MECP2 regulates neuronal receptors and channels / phagocytic vesicle / positive regulation of stress fiber assembly / cardiac muscle contraction / heat shock protein binding / cytoskeleton organization / negative regulation of insulin receptor signaling pathway / endomembrane system / ESR-mediated signaling / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / cellular response to nutrient levels / positive regulation of glycolytic process / cellular response to amino acid starvation / negative regulation of phosphatidylinositol 3-kinase/protein kinase B signal transduction / cellular response to starvation / regulation of signal transduction by p53 class mediator / Regulation of PTEN gene transcription / negative regulation of autophagy / VEGFR2 mediated vascular permeability / post-embryonic development / peptidylprolyl isomerase / TP53 Regulates Metabolic Genes / regulation of actin cytoskeleton organization / response to bacterium / peptidyl-prolyl cis-trans isomerase activity / non-specific protein-tyrosine kinase / positive regulation of translation Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.3 Å | ||||||
 Authors Authors | Maerz, A.M. / Bracher, A. / Hausch, F. | ||||||
 Citation Citation | Journal: Mol.Cell.Biol. / Year: 2013 Title: Large FK506-Binding Proteins Shape the Pharmacology of Rapamycin. Authors: Marz, A.M. / Fabian, A.K. / Kozany, C. / Bracher, A. / Hausch, F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4drh.cif.gz | 205.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4drh.ent.gz | 164.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4drh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dr/4drhftp://data.pdbj.org/pub/pdb/validation_reports/dr/4drh | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4driC  4drjC  1fapS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 15693.832 Da / Num. of mol.: 2 / Fragment: FKBP51 Fk1 domain, UNP RESIDUES 1-140 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: AIG6, FKBP5, FKBP51 / Plasmid: pProEx-Hta / Production host:  #2: Protein | Mass: 11747.374 Da / Num. of mol.: 2 / Fragment: FRB domain, UNP RESIDUES 2025-2114 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: FRAP, FRAP1, FRAP2, MTOR, RAFT1, RAPT1 / Plasmid: pProEx-Hta / Production host: References: UniProt: P42345, non-specific serine/threonine protein kinase #3: Chemical | 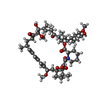  Mass: 914.172 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C51H79NO13 / Comment: antibiotic*YM Mass: 914.172 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C51H79NO13 / Comment: antibiotic*YM#4: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 21 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 21 / Source method: obtained synthetically / Formula: SO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 89 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 89 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.05 Å3/Da / Density % sol: 59.63 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion / pH: 3.5 Details: 0.1M citric acid 2M (NH4)2SO4, pH 3.5, vapor diffusion, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.9814 Å / Beamline: ID29 / Wavelength: 0.9814 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Sep 25, 2008 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9814 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.3→89.803 Å / Num. all: 29100 / Num. obs: 29100 / % possible obs: 99.4 % / Redundancy: 7.4 % / Biso Wilson estimate: 46.27 Å2 / Rmerge(I) obs: 0.081 / Rsym value: 0.081 / Net I/σ(I): 18.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1FAP Resolution: 2.3→20 Å / Cor.coef. Fo:Fc: 0.951 / Cor.coef. Fo:Fc free: 0.934 / WRfactor Rfree: 0.2467 / WRfactor Rwork: 0.215 / Occupancy max: 1 / Occupancy min: 0.4 / FOM work R set: 0.8532 / SU B: 12.061 / SU ML: 0.145 / SU R Cruickshank DPI: 0.2816 / SU Rfree: 0.2124 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.246 / ESU R Free: 0.198 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 121.55 Å2 / Biso mean: 45.9823 Å2 / Biso min: 22.01 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→20 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.359 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
