ジャーナル: PLoS Pathog / 年: 2014 タイトル: The CD27L and CTP1L endolysins targeting Clostridia contain a built-in trigger and release factor. 著者: Matthew Dunne / Haydyn D T Mertens / Vasiliki Garefalaki / Cy M Jeffries / Andrew Thompson / Edward A Lemke / Dmitri I Svergun / Melinda J Mayer / Arjan Narbad / Rob Meijers / 要旨: The bacteriophage ΦCD27 is capable of lysing Clostridium difficile, a pathogenic bacterium that is a major cause for nosocomial infection. A recombinant CD27L endolysin lyses C. difficile in vitro, ...The bacteriophage ΦCD27 is capable of lysing Clostridium difficile, a pathogenic bacterium that is a major cause for nosocomial infection. A recombinant CD27L endolysin lyses C. difficile in vitro, and represents a promising alternative as a bactericide. To better understand the lysis mechanism, we have determined the crystal structure of an autoproteolytic fragment of the CD27L endolysin. The structure covers the C-terminal domain of the endolysin, and represents a novel fold that is identified in a number of lysins that target Clostridia bacteria. The structure indicates endolysin cleavage occurs at the stem of the linker connecting the catalytic domain with the C-terminal domain. We also solved the crystal structure of the C-terminal domain of a slow cleaving mutant of the CTP1L endolysin that targets C. tyrobutyricum. Two distinct dimerization modes are observed in the crystal structures for both endolysins, despite a sequence identity of only 22% between the domains. The dimers are validated to be present for the full length protein in solution by right angle light scattering, small angle X-ray scattering and cross-linking experiments using the cross-linking amino acid p-benzoyl-L-phenylalanine (pBpa). Mutagenesis on residues contributing to the dimer interfaces indicates that there is a link between the dimerization modes and the autocleavage mechanism. We show that for the CTP1L endolysin, there is a reduction in lysis efficiency that is proportional to the cleavage efficiency. We propose a model for endolysin triggering, where the extended dimer presents the inactive state, and a switch to the side-by-side dimer triggers the cleavage of the C-terminal domain. This leads to the release of the catalytic portion of the endolysin, enabling the efficient digestion of the bacterial cell wall.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Clostridium phage phiCTP1 (ファージ)
Clostridium phage phiCTP1 (ファージ) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用


 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード












 PDBj
PDBj 集合体
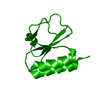
集合体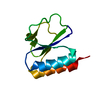

 分子量: 18.015 Da / 分子数: 87 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 87 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析