Entry Database : PDB / ID : 3ztxTitle Aurora kinase selective inhibitors identified using a Taxol-induced checkpoint sensitivity screen. INNER CENTROMERE PROTEIN A SERINE/THREONINE-PROTEIN KINASE 12-A Keywords / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species XENOPUS LAEVIS (African clawed frog)Method / / / Resolution : 1.95 Å Authors Kwiatkowski, N. / Villa, F. / Musacchio, A. / Gray, N. Journal : Acs Chem.Biol. / Year : 2012Title : Selective Aurora Kinase Inhibitors Identified Using a Taxol- Induced Checkpoint Sensitivity Screen.Authors : Kwiatkowski, N. / Deng, X. / Wang, J. / Tan, L. / Villa, F. / Santaguida, S. / Huang, H.C. / Mitchison, T. / Musacchio, A. / Gray, N. History Deposition Jul 12, 2011 Deposition site / Processing site Revision 1.0 Feb 1, 2012 Provider / Type Revision 1.1 Nov 13, 2024 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Structure summary Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_entry_details / pdbx_modification_feature / struct_conn / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _struct_conn.pdbx_leaving_atom_flag / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads














 PDBj
PDBj Assembly
Assembly



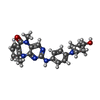
 Mass: 430.502 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H26N6O2
Mass: 430.502 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H26N6O2 Mass: 18.015 Da / Num. of mol.: 285 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 285 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID14-1 / Wavelength: 0.9334
/ Beamline: ID14-1 / Wavelength: 0.9334  Processing
Processing