 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3uth: Crystal structure of Aspergillus fumigatus UDP galactopyranose mu... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3uth | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Aspergillus fumigatus UDP galactopyranose mutase complexed with substrate UDP-Galp in reduced state | ||||||
 Components Components | UDP-galactopyranose mutase | ||||||
 Keywords Keywords | ISOMERASE / Nucleotide binding / Mutase / Flavin adenine dinucleotide binding | ||||||
| Function / homology |  Function and homology information Function and homology informationUDP-galactopyranose mutase / UDP-galactopyranose mutase activity / cell wall organization / nucleotide binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å | ||||||
 Authors Authors | Dhatwalia, R. / Singh, H. / Tanner, J.J. | ||||||
 Citation Citation | Journal: J Biol Chem / Year: 2012 Title: Crystal structures and small-angle x-ray scattering analysis of UDP-galactopyranose mutase from the pathogenic fungus Aspergillus fumigatus. Authors: Richa Dhatwalia / Harkewal Singh / Michelle Oppenheimer / Dale B Karr / Jay C Nix / Pablo Sobrado / John J Tanner /  Abstract: UDP-galactopyranose mutase (UGM) is a flavoenzyme that catalyzes the conversion of UDP-galactopyranose to UDP-galactofuranose, which is a central reaction in galactofuranose biosynthesis. ...UDP-galactopyranose mutase (UGM) is a flavoenzyme that catalyzes the conversion of UDP-galactopyranose to UDP-galactofuranose, which is a central reaction in galactofuranose biosynthesis. Galactofuranose has never been found in humans but is an essential building block of the cell wall and extracellular matrix of many bacteria, fungi, and protozoa. The importance of UGM for the viability of many pathogens and its absence in humans make UGM a potential drug target. Here we report the first crystal structures and small-angle x-ray scattering data for UGM from the fungus Aspergillus fumigatus, the causative agent of aspergillosis. The structures reveal that Aspergillus UGM has several extra secondary and tertiary structural elements that are not found in bacterial UGMs yet are important for substrate recognition and oligomerization. Small-angle x-ray scattering data show that Aspergillus UGM forms a tetramer in solution, which is unprecedented for UGMs. The binding of UDP or the substrate induces profound conformational changes in the enzyme. Two loops on opposite sides of the active site move toward each other by over 10 Å to cover the substrate and create a closed active site. The degree of substrate-induced conformational change exceeds that of bacterial UGMs and is a direct consequence of the unique quaternary structure of Aspergillus UGM. Galactopyranose binds at the re face of the FAD isoalloxazine with the anomeric carbon atom poised for nucleophilic attack by the FAD N5 atom. The structural data provide new insight into substrate recognition and the catalytic mechanism and thus will aid inhibitor design. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3uth.cif.gz | 785.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3uth.ent.gz | 651.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3uth.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ut/3uthftp://data.pdbj.org/pub/pdb/validation_reports/ut/3uth | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3uteSC  3utfC  3utgC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 57128.484 Da / Num. of mol.: 4 / Mutation: K344A, K345A, A429T Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical | ChemComp-FDA /   Mass: 787.566 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H35N9O15P2 Mass: 787.566 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H35N9O15P2#3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: SO4#4: Chemical | 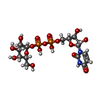  Mass: 566.302 Da / Num. of mol.: 2 Mass: 566.302 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C15H24N2O17P2 #5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 659 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 659 / Source method: isolated from a natural source / Formula: H2OSequence details | AUTHOR STATES THAT A429T IS AN UNINTENDED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.89 Å3/Da / Density % sol: 74.84 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 4.5 Details: 1.5M ammonium sulfate, 0.1M sodium acetate, 5mM L-cysteine, 0.5mM THP , pH 4.5, VAPOR DIFFUSION, SITTING DROP, temperature 298.0K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 24-ID-C / Wavelength: 0.97949 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Nov 16, 2011 Details: Cryogenically-cooled single crystal Si(220) side bounce monochromator. Optional Si(311) to achive 13.474 keV. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Cryo-Cooled double crystal. / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97949 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.25→48.807 Å / Num. all: 207614 / Num. obs: 207614 / % possible obs: 97.3 % / Redundancy: 8 % / Rsym value: 0.097 / Net I/σ(I): 13.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3UTE Resolution: 2.25→48.807 Å / Occupancy max: 1 / Occupancy min: 0 / FOM work R set: 0.8717 / SU ML: 0.32 / σ(F): 0 / Phase error: 20 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 1.06 Å / VDW probe radii: 1.3 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 30.824 Å2 / ksol: 0.352 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 125.46 Å2 / Biso mean: 35.5875 Å2 / Biso min: 12.79 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.25→48.807 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 10
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
