 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3t74 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Thermolysin In Complex With UBTLN27 | ||||||
 Components Components | Thermolysin | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / protease / metalloprotease / hydrolysis of peptide bonds / phosphoramidon / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationthermolysin / metalloendopeptidase activity / proteolysis / extracellular region / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.28 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.28 Å | ||||||
 Authors Authors | Biela, A. / Heine, A. / Klebe, G. | ||||||
 Citation Citation | Journal: Chemmedchem / Year: 2012 Title: Water makes the difference: rearrangement of water solvation layer triggers non-additivity of functional group contributions in protein-ligand binding. Authors: Biela, A. / Betz, M. / Heine, A. / Klebe, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3t74.cif.gz | 163 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3t74.ent.gz | 127.6 KB | Display | PDB format |
| PDBx/mmJSON format | 3t74.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3t74_validation.pdf.gz | 962.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3t74_full_validation.pdf.gz | 963.4 KB | Display | |
| Data in XML | 3t74_validation.xml.gz | 19.2 KB | Display | |
| Data in CIF | 3t74_validation.cif.gz | 30.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/t7/3t74ftp://data.pdbj.org/pub/pdb/validation_reports/t7/3t74 | HTTPS FTP |
-Related structure data
| Related structure data |  3t73C  3t8fC  3t8gC  8tlnS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




























-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 34360.336 Da / Num. of mol.: 1 / Fragment: mature form (UNP residues 233-548) / Source method: isolated from a natural source / Source: (natural) |
|---|
-Non-polymers , 6 types, 513 molecules 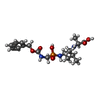





| #2: Chemical | ChemComp-UBY /  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 429.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H28N3O7P Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 429.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H28N3O7PReferences: N-[(S)-({[(BENZYLOXY)CARBONYL]AMINO}METHYL)(HYDROXY)PHOSPHORYL]-L-LEUCYL-L-ALANINE | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #3: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3#4: Chemical | ChemComp-DMS /  Mass: 78.133 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM#5: Chemical | ChemComp-ZN / |  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn#6: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 499 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.36 Å3/Da / Density % sol: 47.87 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 50 mM Tris, 1.9 M cesium chloride, 50% DMSO, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.2 / Wavelength: 0.91841 Å / Beamline: 14.2 / Wavelength: 0.91841 Å |
| Detector | Type: RAYONIX MX-225 / Detector: CCD / Date: Jun 25, 2010 / Details: Collimating Mirror |
| Radiation | Monochromator: Double Crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91841 Å / Relative weight: 1 |
| Reflection | Resolution: 1.28→50 Å / Num. all: 83375 / Num. obs: 83375 / % possible obs: 97.3 % / Redundancy: 5.7 % / Rsym value: 0.052 / Net I/σ(I): 30.4 |
| Reflection shell | Resolution: 1.28→1.3 Å / Redundancy: 5.7 % / Mean I/σ(I) obs: 5.3 / Num. unique all: 4026 / Rsym value: 0.326 / % possible all: 95.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 8TLN Resolution: 1.28→22.797 Å / SU ML: 0.1 / Cross valid method: FREE R / σ(F): 0 / Phase error: 9.63 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.77 Å / VDW probe radii: 0.9 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 53.669 Å2 / ksol: 0.399 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.28→22.797 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
