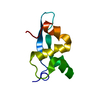
SIGNALING PROTEIN RECEPTOR / human / type I interferons / receptor chain / IFNAR2 / fibronectin type III module / part of type I interferon receptor chain / interferon / extracellular space
Function / homology
Function and homology information
type I interferon receptor activity / type I interferon binding / JAK pathway signal transduction adaptor activity / response to interferon-beta / response to interferon-alpha / type I interferon-mediated signaling pathway / cytokine binding / Regulation of IFNA/IFNB signaling / cell surface receptor signaling pathway via JAK-STAT / cellular response to interferon-beta ...type I interferon receptor activity / type I interferon binding / JAK pathway signal transduction adaptor activity / response to interferon-beta / response to interferon-alpha / type I interferon-mediated signaling pathway / cytokine binding / Regulation of IFNA/IFNB signaling / cell surface receptor signaling pathway via JAK-STAT / cellular response to interferon-beta / cellular response to virus / Evasion by RSV of host interferon responses / response to virus / Interferon alpha/beta signaling / defense response to virus / Potential therapeutics for SARS / cell surface receptor signaling pathway / signaling receptor complex / protein kinase binding / SARS-CoV-2 activates/modulates innate and adaptive immune responses / : / extracellular region / plasma membrane Similarity search - Function
Interferon/interleukin receptor domain / Interferon-alpha/beta receptor, fibronectin type III / : / Tissue factor / Fibronectin type III / Fibronectin type III superfamily / Immunoglobulin-like fold / Immunoglobulins / Immunoglobulin-like / Sandwich / Mainly Beta Similarity search - Domain/homology
Mass: 18.015 Da / Num. of mol.: 35 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 3.07 Å3/Da / Density % sol: 59.99 %
Crystal grow
Temperature: 293 K / pH: 8.5 Details: 31% (w/v) PEG 4000, 200 mM Li2SO4, 100 mM Tris pH 8.5, 5% (v/v) MPD, 10 mM Na acetate, 1.5% (w/v) PEG8000, VAPOR DIFFUSION, HANGING DROP, temperature 293K
Type: MARMOSAIC 325 mm CCD / Detector: CCD / Date: Oct 29, 2009
Radiation
Monochromator: DOUBLE CRYSTAL MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.9791 Å / Relative weight: 1
Reflection
Resolution: 2.6→19.925 Å / Num. obs: 26344 / % possible obs: 99.4 % / Redundancy: 5.6 % / Biso Wilson estimate: 45.4 Å2 / Rsym value: 9 / Net I/σ(I): 16.3
Reflection shell
Resolution: 2.6→2.7 Å / Redundancy: 5.2 % / Mean I/σ(I) obs: 2.4 / Rsym value: 66.4 / % possible all: 99.2
-
Processing
Software
Name
Classification
Blu-Ice
datacollection
autoSHARP
phasing
PHENIX
refinement
XDS
datareduction
XSCALE
datascaling
Refinement
Method to determine structure: SAD / Resolution: 2.6→19.925 Å / σ(F): 1.43 / Stereochemistry target values: ML Details: ANOMALOUS SCATTERER GROUP USED FOR ATOM SE WITH FP = -3.6227 AND FDP = 5.4465
Rfactor
Num. reflection
% reflection
Rfree
0.27
1329
5.05 %
Rwork
0.218
-
-
obs
-
26342
99.6 %
Solvent computation
Shrinkage radii: 0.83 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 45.3 Å2 / ksol: 0.34 e/Å3
Displacement parameters
Biso mean: 51.2 Å2
Baniso -1
Baniso -2
Baniso -3
1-
8.8 Å2
0 Å2
-0 Å2
2-
-
8.8 Å2
0 Å2
3-
-
-
-17.59 Å2
Refinement step
Cycle: LAST / Resolution: 2.6→19.925 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
1950
0
2
35
1987
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.017
2001
X-RAY DIFFRACTION
f_angle_d
1.22
2716
X-RAY DIFFRACTION
f_dihedral_angle_d
17.257
735
X-RAY DIFFRACTION
f_chiral_restr
0.084
324
X-RAY DIFFRACTION
f_plane_restr
0.007
342
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
2.6-2.7039
0.3817
147
0.3029
2769
X-RAY DIFFRACTION
99
2.7039-2.8266
0.2869
143
0.265
2771
X-RAY DIFFRACTION
99
2.8266-2.9752
0.3157
150
0.2657
2753
X-RAY DIFFRACTION
99
2.9752-3.1609
0.259
152
0.2449
2783
X-RAY DIFFRACTION
100
3.1609-3.4038
0.302
145
0.2179
2793
X-RAY DIFFRACTION
100
3.4038-3.7443
0.3145
153
0.1996
2794
X-RAY DIFFRACTION
100
3.7443-4.2815
0.23
145
0.1928
2769
X-RAY DIFFRACTION
100
4.2815-5.3766
0.2076
147
0.1611
2778
X-RAY DIFFRACTION
100
5.3766-19.9252
0.2699
147
0.2498
2805
X-RAY DIFFRACTION
100
Refinement TLS params.
Method: refined / Origin x: 25.2747 Å / Origin y: -26.4021 Å / Origin z: 0.5411 Å
11
12
13
21
22
23
31
32
33
T
0.117 Å2
-0.0234 Å2
0.0032 Å2
-
0.1367 Å2
-0.0365 Å2
-
-
0.1371 Å2
L
0.7434 °2
0.301 °2
-0.6159 °2
-
0.2268 °2
-0.0223 °2
-
-
1.0209 °2
S
-0.0554 Å °
0.0162 Å °
0.005 Å °
0.043 Å °
0.0463 Å °
-0.0223 Å °
-0.0029 Å °
-0.0412 Å °
-0.0014 Å °
Refinement TLS group
Selection details: all
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj



 Assembly
Assembly



 Trichoplusia ni (cabbage looper) / References: UniProt: P48551
Trichoplusia ni (cabbage looper) / References: UniProt: P48551
 Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 18.015 Da / Num. of mol.: 35 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 35 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL9-2 / Wavelength: 0.9791
/ Beamline: BL9-2 / Wavelength: 0.9791  Processing
Processing