 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3rdc | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human Cyclophilin D Complexed with an Inhibitor | ||||||
 Components Components | Peptidyl-prolyl cis-trans isomerase F, mitochondrial | ||||||
 Keywords Keywords | ISOMERASE/ISOMERASE INHIBITOR / beta barrel / prolyl cis/trans isomerase / mitochondria / inhibitor / ISOMERASE-ISOMERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology information: / mitochondrial outer membrane permeabilization involved in programmed cell death / regulation of mitochondrial membrane permeability involved in programmed necrotic cell death / skeletal muscle fiber differentiation / mitochondrial permeability transition pore complex / cellular response to arsenic-containing substance / mitochondrial depolarization / negative regulation of oxidative phosphorylation / regulation of mitochondrial membrane permeability / cyclosporin A binding ...: / mitochondrial outer membrane permeabilization involved in programmed cell death / regulation of mitochondrial membrane permeability involved in programmed necrotic cell death / skeletal muscle fiber differentiation / mitochondrial permeability transition pore complex / cellular response to arsenic-containing substance / mitochondrial depolarization / negative regulation of oxidative phosphorylation / regulation of mitochondrial membrane permeability / cyclosporin A binding / negative regulation of release of cytochrome c from mitochondria / necroptotic process / negative regulation of intrinsic apoptotic signaling pathway / apoptotic mitochondrial changes / cellular response to calcium ion / response to ischemia / peptidylprolyl isomerase / peptidyl-prolyl cis-trans isomerase activity / cellular response to hydrogen peroxide / protein folding / mitochondrial matrix / negative regulation of apoptotic process / mitochondrion / membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.94 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.94 Å | ||||||
 Authors Authors | Colliandre, L. / Ahmed-Belkacem, H. / Bessin, Y. / Pawlotsky, J.M. / Guichou, J.F. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2015 Title: Combining 'dry' co-crystallization and in situ diffraction to facilitate ligand screening by X-ray crystallography. Authors: Gelin, M. / Delfosse, V. / Allemand, F. / Hoh, F. / Sallaz-Damaz, Y. / Pirocchi, M. / Bourguet, W. / Ferrer, J.L. / Labesse, G. / Guichou, J.F. #1: Journal: Nat Commun / Year: 2016Title: Fragment-based discovery of a new family of non-peptidic small-molecule cyclophilin inhibitors with potent antiviral activities. Authors: Ahmed-Belkacem, A. / Colliandre, L. / Ahnou, N. / Nevers, Q. / Gelin, M. / Bessin, Y. / Brillet, R. / Cala, O. / Douguet, D. / Bourguet, W. / Krimm, I. / Pawlotsky, J.M. / Guichou, J.F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3rdc.cif.gz | 50.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3rdc.ent.gz | 34.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3rdc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rd/3rdcftp://data.pdbj.org/pub/pdb/validation_reports/rd/3rdc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4xldC  4xn6C  4xncC  4xneC  4xoyC  4xozC  4xp0C  4xp2C  4xp3C  4xrjC  4xrlC  4zscC  4zsdC  2bitS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |

























-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 17870.400 Da / Num. of mol.: 1 / Fragment: UNP residues 43-207 / Mutation: K175I Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PPIF, CYP3 / Production host:  |
|---|---|
| #2: Chemical | ChemComp-EA4 / 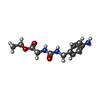  Mass: 251.282 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H17N3O3 Mass: 251.282 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H17N3O3 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 223 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 223 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.94 Å3/Da / Density % sol: 36.56 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.3 Details: 30% PEG4000, pH 7.3, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: AREA DETECTOR / Date: Oct 12, 2008 |
| Radiation | Monochromator: yale mirror / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.93→28.2 Å / Num. all: 10623 / Num. obs: 10358 / % possible obs: 97.5 % / Observed criterion σ(I): 9.1 / Redundancy: 4.1 % / Rsym value: 0.059 |
| Reflection shell | Resolution: 1.93→1.987 Å / Redundancy: 4.8 % / Mean I/σ(I) obs: 3.5 / Rsym value: 0.188 / % possible all: 84 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2BIT Resolution: 1.94→25.79 Å / Cor.coef. Fo:Fc: 0.908 / Cor.coef. Fo:Fc free: 0.853 / SU B: 4.259 / SU ML: 0.13 / Cross valid method: THROUGHOUT / ESU R: 0.232 / ESU R Free: 0.201 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.287 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.94→25.79 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.937→1.987 Å / Total num. of bins used: 20
|
