 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3opz: Crystal structure of trans-sialidase in complex with the Fab frag... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3opz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of trans-sialidase in complex with the Fab fragment of a neutralizing monoclonal IgG antibody | ||||||
 Components Components |
| ||||||
 Keywords Keywords | hydrolase/immune system / six-bladed beta-propeller neuraminidase immunoglobulin domain / viral protein-immune system complex / hydrolase-immune system complex | ||||||
| Function / homology |  Function and homology information Function and homology informationganglioside catabolic process / oligosaccharide catabolic process / exo-alpha-sialidase activity / membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |   | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 3.4 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 3.4 Å | ||||||
 Authors Authors | Larrieux, N. / Muia, R. / Campetella, O. / Buschiazzo, A. | ||||||
 Citation Citation | Journal: Plos Pathog. / Year: 2012 Title: Trypanosoma cruzi trans-sialidase in complex with a neutralizing antibody: structure/function studies towards the rational design of inhibitors. Authors: Buschiazzo, A. / Muia, R. / Larrieux, N. / Pitcovsky, T. / Mucci, J. / Campetella, O. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3opz.cif.gz | 1.2 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3opz.ent.gz | 1016.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3opz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/op/3opzftp://data.pdbj.org/pub/pdb/validation_reports/op/3opz | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Refine code: 1
NCS ensembles :
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | The biological assembly is a heterotrimer, with three trimers in the ASU (chains A-H-L, B-I-M and C-J-N). Each heterotrimers corresponds to a stable binary complex between trans-sialidase (e.g. chain A) and a Fab IgG fragment, itself a heterodimer (chains H and L). |
-Components
-Protein , 1 types, 3 molecules ABC
| #1: Protein | Mass: 71389.258 Da / Num. of mol.: 3 Mutation: N59F,S263T,R477H,V485L,S496K,V497G,E521K,E559V,D594G,I598D,H600R Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|
-Antibody , 2 types, 6 molecules HIJLMN
| #2: Antibody | Mass: 23869.732 Da / Num. of mol.: 3 / Source method: isolated from a natural source Details: monoclonal antibody was obtained from hybridomas (after fusion of mice splenocytes with Sp2/0 cells), and the Fab fragment obtained by standard papain digestion Source: (natural) #3: Antibody | Mass: 23382.764 Da / Num. of mol.: 3 / Source method: isolated from a natural source Details: monoclonal antibody was obtained from hybridomas (after fusion of mice splenocytes with Sp2/0 cells), and the Fab fragment obtained by standard papain digestion Source: (natural) |
|---|
-Non-polymers , 3 types, 45 molecules 
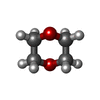

| #4: Chemical | ChemComp-NA /  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na | ||
|---|---|---|---|
| #5: Chemical |  Mass: 88.105 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H8O2 Mass: 88.105 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H8O2#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 42 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|---|
| Sequence details | AUTHORS STATE THAT UNIPROT ENTRY Q26966 HAS FOUR SEQUENCE ERRORS. THESE DISCREPANCIES HAVE BEEN ...AUTHORS STATE THAT UNIPROT ENTRY Q26966 HAS FOUR SEQUENCE ERRORS. THESE DISCREPANC |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.62 Å3/Da / Density % sol: 66.04 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: 0.1 M Bicina, 10 % PEG 20000, 4% 1,4-dioxano, pH 8.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 108 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.5418 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Apr 27, 2009 / Details: Varimax HF multilayer mirrors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: multilayer mirrors (Varimax HF) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 3.4→154.273 Å / Num. all: 68611 / Num. obs: 68611 / % possible obs: 99.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.6 % / Rsym value: 0.19 / Net I/σ(I): 6.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Rfactor: 35.39 / Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 3.4→29.79 Å / Cor.coef. Fo:Fc: 0.934 / Cor.coef. Fo:Fc free: 0.891 / WRfactor Rfree: 0.1811 / WRfactor Rwork: 0.1456 / Occupancy max: 1 / Occupancy min: 1 / FOM work R set: 0.8948 / SU B: 44.979 / SU ML: 0.316 / SU R Cruickshank DPI: 0.3886 / SU Rfree: 0.4221 / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.422 / Stereochemistry target values: Engh & Huber / Details: U VALUES : WITH TLS COMPONENT INCLUDED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 203.88 Å2 / Biso mean: 59.3104 Å2 / Biso min: 2 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.4→29.79 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.4→3.487 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
