 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3g15: Crystal structure of human choline kinase alpha in complex with h... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3g15 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human choline kinase alpha in complex with hemicholinium-3 and ADP | |||||||||
 Components Components | Choline kinase alpha | |||||||||
 Keywords Keywords | TRANSFERASE / NON-PROTEIN KINASE / CHOLINE KINASE / STRUCTURAL GENOMICS Consortium / SGC / hemicholinium-3 / Kinase | |||||||||
| Function / homology |  Function and homology information Function and homology informationethanolamine kinase / choline kinase / Synthesis of PE / ethanolamine kinase activity / choline kinase activity / CDP-choline pathway / lipid droplet disassembly / phosphatidylethanolamine biosynthetic process / phosphatidylcholine biosynthetic process / cholinesterase activity ...ethanolamine kinase / choline kinase / Synthesis of PE / ethanolamine kinase activity / choline kinase activity / CDP-choline pathway / lipid droplet disassembly / phosphatidylethanolamine biosynthetic process / phosphatidylcholine biosynthetic process / cholinesterase activity / lipid transport / Synthesis of PC / cellular response to glucose starvation / lipid droplet / lipid metabolic process / protein tyrosine kinase activity / protein homodimerization activity / ATP binding / cytoplasm / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.7 Å | |||||||||
 Authors Authors | Hong, B.S. / Tempel, W. / Rabeh, W.M. / MacKenzie, F. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Weigelt, J. / Bochkarev, A. / Park, H.W. / Structural Genomics Consortium (SGC) | |||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2010 Title: Crystal structures of human choline kinase isoforms in complex with hemicholinium-3: single amino acid near the active site influences inhibitor sensitivity. Authors: Hong, B.S. / Allali-Hassani, A. / Tempel, W. / Finerty, P.J. / Mackenzie, F. / Dimov, S. / Vedadi, M. / Park, H.W. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3g15.cif.gz | 165.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3g15.ent.gz | 124.2 KB | Display | PDB format |
| PDBx/mmJSON format | 3g15.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g1/3g15ftp://data.pdbj.org/pub/pdb/validation_reports/g1/3g15 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3fegC  3lq3C  2i7qS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 46856.477 Da / Num. of mol.: 2 / Fragment: UNP residues 75-457 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CHKA, CHK, CKI / Plasmid: pET28aLIC / Production host:  |
|---|
-Non-polymers , 6 types, 355 molecules 

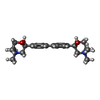



| #2: Chemical |  Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#3: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#4: Chemical |  Mass: 414.538 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H34N2O4 Mass: 414.538 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H34N2O4#5: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#6: Chemical | ChemComp-UNX /  Num. of mol.: 21 / Source method: obtained synthetically Num. of mol.: 21 / Source method: obtained synthetically#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 324 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 47.01 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion / pH: 7.5 Details: 25% PEG-3350, 0.2M lithium sulfate, 0.1M HEPES. Crystallization sample buffer: 0.01M TRIS pH 8.0, 0.5M sodium chloride, 0.005M magnesium chloride, 0.01M DTT, 0.003M hemicholinium-3, 0.005M ...Details: 25% PEG-3350, 0.2M lithium sulfate, 0.1M HEPES. Crystallization sample buffer: 0.01M TRIS pH 8.0, 0.5M sodium chloride, 0.005M magnesium chloride, 0.01M DTT, 0.003M hemicholinium-3, 0.005M ADP., vapor diffusion, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 23-ID-B / Wavelength: 0.96863 Å / Beamline: 23-ID-B / Wavelength: 0.96863 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MAR scanner 300 mm plate / Detector: IMAGE PLATE / Date: Feb 14, 2008 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.96863 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.7→50 Å / Num. obs: 93657 / % possible obs: 97 % / Redundancy: 6.5 % / Rmerge(I) obs: 0.069 / Χ2: 1.585 / Net I/σ(I): 11.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2I7Q Resolution: 1.7→30 Å / Cor.coef. Fo:Fc: 0.947 / Cor.coef. Fo:Fc free: 0.922 / WRfactor Rfree: 0.25 / WRfactor Rwork: 0.213 / SU B: 2.363 / SU ML: 0.079 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.112 / ESU R Free: 0.113 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. HEMICHOLINIUM-3 RESTRAINTS WERE PREPARED BY THE PRODRG SERVER.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.163 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→30 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
|
