 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3d0u | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Lysine Riboswitch Bound to Lysine | ||||||
 Components Components | Lysine Riboswitch RNA | ||||||
 Keywords Keywords | RNA / RNA-ligand complex / riboswitch | ||||||
| Function / homology | IRIDIUM HEXAMMINE ION / LYSINE / RNA / RNA (> 10) / RNA (> 100) Function and homology information Function and homology information | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.8 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.8 Å | ||||||
 Authors Authors | Garst, A.D. / Heroux, A. / Rambo, R.P. / Batey, R.T. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2008 Title: Crystal structure of the lysine riboswitch regulatory mRNA element. Authors: Garst, A.D. / Heroux, A. / Rambo, R.P. / Batey, R.T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3d0u.cif.gz | 100.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3d0u.ent.gz | 73 KB | Display | PDB format |
| PDBx/mmJSON format | 3d0u.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/d0/3d0uftp://data.pdbj.org/pub/pdb/validation_reports/d0/3d0u | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj





























- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: RNA chain | Mass: 52465.289 Da / Num. of mol.: 1 / Fragment: Ligand binding domain / Source method: obtained synthetically / Details: sequence occurs naturally in Thermotoga Maritima | ||||
|---|---|---|---|---|---|
| #2: Chemical | 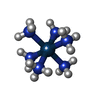  Mass: 294.400 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: H18IrN6 Mass: 294.400 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: H18IrN6#3: Chemical | ChemComp-LYS / | 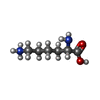  Type: L-peptide linking / Mass: 147.195 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15N2O2 Type: L-peptide linking / Mass: 147.195 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15N2O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 45 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 45 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.64 Å3/Da / Density % sol: 73.5 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 303 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 2 M Li2SO4, 5 mM MgCl2, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 303K | ||||||||||||||||||||
| Components of the solutions |
|
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X29A / Wavelength: 1.105 Å / Beamline: X29A / Wavelength: 1.105 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Jan 28, 2008 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Si 111 CHANNEL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.105 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Redundancy: 5 % / Av σ(I) over netI: 13.5 / Number: 122605 / Rmerge(I) obs: 0.084 / Χ2: 2.35 / D res high: 2.7 Å / D res low: 40 Å / Num. obs: 24657 / % possible obs: 95.6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.7→40 Å / Num. obs: 24657 / % possible obs: 95.6 % / Redundancy: 5 % / Rmerge(I) obs: 0.084 / Χ2: 2.353 / Net I/σ(I): 13.5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Resolution: 2.7→2.8 Å / Redundancy: 2.2 % / Rmerge(I) obs: 0.403 / Num. unique all: 1601 / Χ2: 0.404 / % possible all: 61.4 |
-Phasing
| Phasing | Method: SAD |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.8→32.619 Å / FOM work R set: 0.862 / σ(F): 0.41 / Stereochemistry target values: MLHL
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 8.453 Å2 / ksol: 0.208 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 16.66 Å2 / Biso mean: 54.61 Å2 / Biso min: 549.57 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→32.619 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 13
|
