 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3bls | ||||||
|---|---|---|---|---|---|---|---|
| Title | AMPC BETA-LACTAMASE FROM ESCHERICHIA COLI | ||||||
 Components Components | AMPC BETA-LACTAMASE | ||||||
 Keywords Keywords | CEPHALOSPORINASE / BETA-LACTAMASE / SERINE HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationantibiotic catabolic process / beta-lactamase activity / beta-lactamase / outer membrane-bounded periplasmic space / response to antibiotic Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR SUBSTITUTION / Resolution: 2.3 Å X-RAY DIFFRACTION / MOLECULAR SUBSTITUTION / Resolution: 2.3 Å | ||||||
 Authors Authors | Usher, K.C. / Shoichet, B.K. / Remington, S.J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1998 Title: Three-dimensional structure of AmpC beta-lactamase from Escherichia coli bound to a transition-state analogue: possible implications for the oxyanion hypothesis and for inhibitor design. Authors: Usher, K.C. / Blaszczak, L.C. / Weston, G.S. / Shoichet, B.K. / Remington, S.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3bls.cif.gz | 150.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3bls.ent.gz | 118.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3bls.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bl/3blsftp://data.pdbj.org/pub/pdb/validation_reports/bl/3bls | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2blsSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.99824, 0.058222, -0.011253), Vector: |
-Components
| #1: Protein | Mass: 39587.922 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | 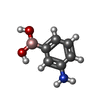  Mass: 136.944 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8BNO2 Mass: 136.944 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8BNO2#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 107 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 107 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8.7 Details: PROTEIN WAS CRYSTALLIZED FROM 1.7M NA/K PHOSPHATE, PH 8.7, COCRYSTALLIZED IN THE PRESENCE OF MAPB INHIBITOR. LARGER CRYSTALS WERE GROWN BY MICRO-SEEDING TECHNIQUE. | |||||||||||||||
| Crystal | *PLUS | |||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop / Details: used to seeding / PH range low: 8.7 / PH range high: 8.5 | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: XUONG-HAMLIN MULTIWIRE / Detector: AREA DETECTOR / Date: Nov 1, 1995 / Details: COLLIMATOR |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→25 Å / Num. obs: 35225 / % possible obs: 95.5 % / Observed criterion σ(I): 0 / Redundancy: 3.2 % / Biso Wilson estimate: 18.6 Å2 / Rmerge(I) obs: 0.088 / Net I/σ(I): 10.1 |
| Reflection shell | Resolution: 2.3→2.48 Å / Redundancy: 2 % / Rmerge(I) obs: 0.216 / Mean I/σ(I) obs: 2.7 / % possible all: 88.3 |
| Reflection | *PLUS Num. all: 36886 / Num. measured all: 114023 |
| Reflection shell | *PLUS % possible obs: 88.3 % / Num. possible: 7324 / Num. unique obs: 6468 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR SUBSTITUTION Starting model: PDB ENTRY 2BLS Resolution: 2.3→25 Å / Isotropic thermal model: TNT BCORREL / σ(F): 0 / Stereochemistry target values: TNT PROTGEO
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: BABINET SCALING / Bsol: 531 Å2 / ksol: 0.99 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→25 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: TNT / Version: 5F / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
