- PDB-3b7m: Crystal structure of a meso-active thermo-stable cellulase (MT Ce... -
+
Open data
ID or keywords:
Loading...
-
Basic information
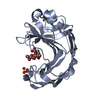
Entry
Database: PDB / ID: 3b7m
Title
Crystal structure of a meso-active thermo-stable cellulase (MT Cel12A) derived by making non-contiguous mutations in the active surface of the Cel12A cellulase of Rhodothermus marinus
SEQUENCE The sequence is based on PDB_ID 1H0B (gi:33356963). THE FOLLOWING MUTATIONS WERE MADE TO ...SEQUENCE The sequence is based on PDB_ID 1H0B (gi:33356963). THE FOLLOWING MUTATIONS WERE MADE TO TRANSFORM THE RHODOTHERMUS MARINUS CEL12A (PDB_ID 1H0B) TO MESO -ACTIVE THERMO-STABLE CEL12A, NAMED MT CEL12A (PDB _ID 3B7M). UNLESS OTHERWISE MENTIONED, THE MUTATIONS REPLACE RESIDUES CONSTITUTING THE ACTIVE SURFACE (CELLULOSE-BINDING AND CATALYTIC SITE RESIDUES)OF 1H0B (RHODOTHERMUS MARINUS CEL12A) BY RESIDUES OCCURRING AT STRUCTURALLY EQUIVALENT POSITIONS ON THE ACTIVE SURFACE OF 1OA2 (TRICHODERMA REESEI CEL12A), TO PRODUCE MT CEL12A. 1H0B SEQUENCE 3B7M SEQUENCE A1 MET A1 SER A8 ARG A8 GLN A11 ALA A11 THR A13 ASP A13 THR A20 ARG A20 THR A22 ILE A22 SER A49 ASP A49 GLU (SEQUENCE ACCORDING TO OUR CLONE) A61 ALA A61 ASN A63 TYR A63 GLN A65 GLY A65 ALA LOOP A66 CYS TO A77 LEU LOOP A66 ILE TO A68 GLN A108 TRP A99 PHE A110 SER A101 ALA A111 PRO A102 ALA A112 VAL A103 ASN A113 THR A104 PRO LOOP A115 SER TO A118 GLY LOOP A106 HIS TO A108 THR A122 GLY A112 ASP A131 TRP A121 LYS A134 GLY A124 ASP A138 GLY A128 ILE A159 TRP A149 ASN A160 ASP A150 GLY BETWEEN A160 ASP AND A161 TRP A151 ALA IS INSERTED A161 TRP A152 MET A163 TYR A154 VAL A165 ALA A156 SER A167 ARG A158 VAL A176 SER A167 THR (ADOPTED FROM APPL MICROBIOL BIOTECHNOL. V55, P578) A200 HIS A191 LEU A201 ALA A192 SER A203 GLU A194 GLN A209 TRP A200 PHE A210 GLU A201 THR
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Rhodothermus marinus (bacteria)
Rhodothermus marinus (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj





 Assembly
Assembly




 Mass: 18.015 Da / Num. of mol.: 293 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 293 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing