 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-366d: 1.3 A STRUCTURE DETERMINATION OF THE D(CG(5-BRU)ACG)2/6-BROMO-9-A... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 366d | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | 1.3 A STRUCTURE DETERMINATION OF THE D(CG(5-BRU)ACG)2/6-BROMO-9-AMINO-DACA COMPLEX | ||||||||||||||||||
 Components Components | DNA (5'-D(* Keywords KeywordsDNA / DRUG INTERCALATION / MAJOR GROOVE BINDING | Function / homology | Chem-DA6 / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO / Resolution: 1.3 Å X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO / Resolution: 1.3 Å  Authors AuthorsTodd, A.K. / Adams, A. / Thorpe, J.H. / Denny, W.A. / Cardin, C.J. |  CitationJournal: J.Med.Chem. / Year: 1999 CitationJournal: J.Med.Chem. / Year: 1999Title: Major groove binding and 'DNA-induced' fit in the intercalation of a derivative of the mixed topoisomerase I/II poison N-(2-(dimethylamino)ethyl)acridine-4-carboxamide (DACA) into DNA: X-ray ...Title: Major groove binding and 'DNA-induced' fit in the intercalation of a derivative of the mixed topoisomerase I/II poison N-(2-(dimethylamino)ethyl)acridine-4-carboxamide (DACA) into DNA: X-ray structure complexed to d(CG(5-BrU)ACG)2 at 1.3-A resolution. Authors: Todd, A.K. / Adams, A. / Thorpe, J.H. / Denny, W.A. / Wakelin, L.P. / Cardin, C.J. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 366d.cif.gz | 24.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb366d.ent.gz | 16.9 KB | Display | PDB format |
| PDBx/mmJSON format | 366d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/66/366dftp://data.pdbj.org/pub/pdb/validation_reports/66/366d | HTTPS FTP |
|---|
-Related structure data






-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 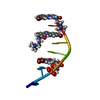
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: DNA chain | Mass: 1874.087 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: COMPLEXED WITH 6-BROMO-9-AMINO-N-ETHYL(DIAMINOMETHYL)ACRIDINE-4-CARBOXAMIDE | ||
|---|---|---|---|
| #2: Chemical | 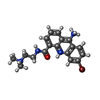  Mass: 389.290 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H21BrN4O Mass: 389.290 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H21BrN4O#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 30 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 30 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.74 Å3/Da / Density % sol: 54 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 6.5 / Details: pH 6.5, VAPOR DIFFUSION, HANGING DROP | |||||||||||||||||||||||||||||||||||
| Components of the solutions |
| |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 285 K / Method: vapor diffusion, sitting drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ELETTRA  / Beamline: 5.2R / Wavelength: 1 / Beamline: 5.2R / Wavelength: 1 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 14, 1997 / Details: MIRRORS |
| Radiation | Monochromator: SI 111 CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.3→8 Å / Num. obs: 9362 / % possible obs: 95.3 % / Redundancy: 3.91 % / Rmerge(I) obs: 0.06 |
| Reflection | *PLUS Num. obs: 9306 / Num. measured all: 31514 / Rmerge(I) obs: 0.0675 |
| Reflection shell | *PLUS Highest resolution: 1.3 Å / Lowest resolution: 1.4 Å / % possible obs: 37 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: AB INITIO / Resolution: 1.3→8 Å / Num. parameters: 3275 / Num. restraintsaints: 8013
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 1 / Occupancy sum non hydrogen: 401.5 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.3→8 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-96 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor all: 0.1468 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
