Mass: 40421.789 Da / Num. of mol.: 1 / Fragment: INTERNALIN DOMAIN AND B-REPEAT, RESIDUES 36-392 Source method: isolated from a genetically manipulated source Details: THE B-REPEAT (RESIDUES 322-392) IS NOT VISIBLE IN THE ELECTRON DENSITY. Source: (gene. exp.) LISTERIA MONOCYTOGENES (bacteria) / Strain: EGD / Production host: ESCHERICHIA COLI (E. coli) / Strain (production host): BL21 / Variant (production host): CODON PLUS / References: UniProt: P25147, UniProt: P0DQD2*PLUS
Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn
Nonpolymer details
ZN ION (ZN): ZN IONS PROBABLY CARRIED OVER FROM ZN CONTAINING SEED SOLUTION. METAL ION VERIFIED BY ...ZN ION (ZN): ZN IONS PROBABLY CARRIED OVER FROM ZN CONTAINING SEED SOLUTION. METAL ION VERIFIED BY ANOMALOUS DIFFERENCE DENSITY.
Sequence details
FIVE N-TERMINAL RESIDUES GPLGS REMAIN AFTER PRESCISSION PROTEASE CLEAVAGE OF THE GST TAG.
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 6.16 Å3/Da / Density % sol: 80 % / Description: NONE
Crystal grow
Temperature: 293 K / Method: vapor diffusion Details: 20 DEG C IN HANGING OR SITTING-DROPS WITH 1.5 UL PROTEIN (5 MG/ML) PLUS 1.5 UL RESERVOIR (0.1 M MES, PH 5.5, 14 % MPD) MICROSEEDED FROM A SIMILAR CONDITION (0.1 M MES PH 6.0, 6.5 % PEG6000 AND 5 MM ZNCL2).
Resolution: 3.2→14.97 Å / Cor.coef. Fo:Fc: 0.949 / Cor.coef. Fo:Fc free: 0.932 / SU B: 27.176 / SU ML: 0.22 / Cross valid method: THROUGHOUT / ESU R: 0.375 / ESU R Free: 0.278 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES WITH TLS ADDED
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.22475
829
5 %
RANDOM
Rwork
0.19713
-
-
-
obs
0.19847
15752
100 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information LISTERIA MONOCYTOGENES (bacteria)
LISTERIA MONOCYTOGENES (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads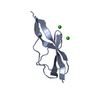






















 PDBj
PDBj






 Assembly
Assembly


 Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn Sample preparation
Sample preparation / Beamline: X12 / Wavelength: 0.98
/ Beamline: X12 / Wavelength: 0.98  Processing
Processing