 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2w2e: 1.15 Angstrom crystal structure of P.pastoris aquaporin, Aqy1, in... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2w2e | ||||||
|---|---|---|---|---|---|---|---|
| Title | 1.15 Angstrom crystal structure of P.pastoris aquaporin, Aqy1, in a closed conformation at pH 3.5 | ||||||
 Components Components | AQUAPORIN PIP2-7 7 | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / YEAST / GATING | ||||||
| Function / homology |  Function and homology information Function and homology informationwater channel activity / endoplasmic reticulum membrane / metal ion binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  KOMAGATAELLA PASTORIS (fungus) KOMAGATAELLA PASTORIS (fungus) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.15 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.15 Å | ||||||
 Authors Authors | Fischer, G. / Kosinska-Eriksson, U. / Aponte-Santamaria, C. / Palmgren, M. / Geijer, C. / Hedfalk, K. / Hohmann, S. / de Groot, B.L. / Neutze, R. / Lindkvist-Petersson, K. | ||||||
 Citation Citation | Journal: Plos Biol. / Year: 2009 Title: Crystal Structure of a Yeast Aquaporin at 1.15 A Reveals a Novel Gating Mechanism.1.15 A Authors: Fischer, G. / Kosinska-Eriksson, U. / Aponte-Santamaria, C. / Palmgren, M. / Geijer, C. / Hedfalk, K. / Hohmann, S. / De Groot, B.L. / Neutze, R. / Lindkvist-Petersson, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2w2e.cif.gz | 126.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2w2e.ent.gz | 98 KB | Display | PDB format |
| PDBx/mmJSON format | 2w2e.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/w2/2w2eftp://data.pdbj.org/pub/pdb/validation_reports/w2/2w2e | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2w1pC  1j4nS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 29932.436 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) KOMAGATAELLA PASTORIS (fungus) / Strain: X33 / References: UniProt: F2QVG4 | ||||
|---|---|---|---|---|---|
| #2: Sugar | ChemComp-BOG / 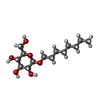  Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 6 Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 6Source method: isolated from a genetically manipulated source Formula: C14H28O6 / Comment: detergent*YM #3: Chemical |   Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 143 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 143 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.43 Å3/Da / Density % sol: 49.4 % / Description: NONE |
|---|---|
| Crystal grow | pH: 3.5 Details: 28% PEG400, 100 MM SODIUM CITRATE PH=3.5, 200 MM LITHIUM SULFATE |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 0.8726 / Beamline: ID23-2 / Wavelength: 0.8726 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Sep 8, 2007 / Details: MIRRORS |
| Radiation | Monochromator: SI 111 CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8726 Å / Relative weight: 1 |
| Reflection | Resolution: 1.15→20 Å / Num. obs: 111455 / % possible obs: 94.7 % / Redundancy: 4.1 % / Rmerge(I) obs: 0.08 / Net I/σ(I): 9.31 |
| Reflection shell | Resolution: 1.15→1.2 Å / Redundancy: 3.3 % / Rmerge(I) obs: 0.73 / Mean I/σ(I) obs: 2.16 / % possible all: 99.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1J4N Resolution: 1.15→20 Å / Num. parameters: 20766 / Num. restraintsaints: 27331 / Cross valid method: FREE R-VALUE / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: H-ATOMS ADDED TO ALL RESIDUES AS A RIDING MODEL, EXCEPT TO THE ONES SURROUNDING THE WATER CHANNEL TO BE ABLE TO OBSERVE DIFFERENCE IN DENSITY. SIDECHAINS OF RESIDUES GLN12, GLU14 AND ARG236 ...Details: H-ATOMS ADDED TO ALL RESIDUES AS A RIDING MODEL, EXCEPT TO THE ONES SURROUNDING THE WATER CHANNEL TO BE ABLE TO OBSERVE DIFFERENCE IN DENSITY. SIDECHAINS OF RESIDUES GLN12, GLU14 AND ARG236 COULD NOT BE OBSERVED WITH SUFFICIENT OCCUPANCY - OCCUPANCY FOR THE NON-OBSERVED ATOMS WAS SET TO 0. SIDE-CHAINS OF RESIDUES ASP77 AND ARG123 COULD ONLY BE OBSERVED WITH PARTIAL OCCUPANCY AND THUS SET TO AN OCCUPANCY OF 0.5. 10 N-TERMINAL AND 6 C-TERMINAL RESIDUES COULD NOT BE OBSERVED.
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 9 / Occupancy sum hydrogen: 1936.39 / Occupancy sum non hydrogen: 2192.03 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.15→20 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
