Mass: 18.015 Da / Num. of mol.: 135 / Source method: isolated from a natural source / Formula: H2O
-
Details
Sequence details
THE NATIVE GLURDELTA2 IS A MEMBRANE PROTEIN. THE CRYSTALLIZED PROTEIN IS THE EXTRACELLULAR LIGAND- ...THE NATIVE GLURDELTA2 IS A MEMBRANE PROTEIN. THE CRYSTALLIZED PROTEIN IS THE EXTRACELLULAR LIGAND-BINDING CORE OF GLURDELTA2. TRANSMEMBRANE REGIONS WERE GENETICALLY REMOVED AND REPLACED WITH A GLY-THR LINKER. CONSEQUENTLY, THE PROTEIN SEQUENCE MATCHES DISCONTINOUSLY WITH THE REFERENCE DATABASE. THE FIRST GLYCINE OF THE SEQUENCE IS A REMNANT OF A TRYPSIN CLEVAGE SITE
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2 Å3/Da / Density % sol: 40 % Description: THE TWO SEPARATE DOMAINS OF THE APO STRUCTURE WERE USED AS SEARCH MODELS AS BINDING OF D-SERINE WAS EXPECTED TO INDUCE CONFORMATIONAL CHANGES.
Crystal grow
Method: vapor diffusion, hanging drop / pH: 6.5 Details: HANGING DROP VAPOR DIFFUSION. 20% PEG4000, 0.1 M CACODYLATE PH 6.5, 0.2 M SODIUM THIOCYANATE
Resolution: 1.74→40.19 Å / Cor.coef. Fo:Fc: 0.954 / Cor.coef. Fo:Fc free: 0.923 / SU B: 2.762 / SU ML: 0.091 / Cross valid method: THROUGHOUT / ESU R: 0.148 / ESU R Free: 0.144 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. RESIDUES 167-171, 215-217 AND 262-265 ARE DISORDERED AND COULD NOT BE MODELED.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.25
1154
5.2 %
RANDOM
Rwork
0.197
-
-
-
obs
0.2
21250
95.3 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj


 Assembly
Assembly

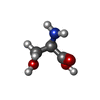



 Type: D-peptide linking / Mass: 105.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO3
Type: D-peptide linking / Mass: 105.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO3 Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
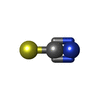
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 58.082 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CNS
Mass: 58.082 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CNS Sample preparation
Sample preparation / Beamline: 14.1 / Wavelength: 0.95373
/ Beamline: 14.1 / Wavelength: 0.95373  Processing
Processing