 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2trm: THE THREE-DIMENSIONAL STRUCTURE OF ASN102 MUTANT OF TRYPSIN. ROLE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2trm | ||||||
|---|---|---|---|---|---|---|---|
| Title | THE THREE-DIMENSIONAL STRUCTURE OF ASN102 MUTANT OF TRYPSIN. ROLE OF ASP102 IN SERINE PROTEASE CATALYSIS | ||||||
 Components Components | TRYPSIN | ||||||
 Keywords Keywords | HYDROLASE (SERINE PROTEINASE) | ||||||
| Function / homology |  Function and homology information Function and homology informationAntimicrobial peptides / Alpha-defensins / Activation of Matrix Metalloproteinases / Neutrophil degranulation / collagen catabolic process / trypsin / digestion / response to nutrient / serine-type endopeptidase activity / calcium ion binding ...Antimicrobial peptides / Alpha-defensins / Activation of Matrix Metalloproteinases / Neutrophil degranulation / collagen catabolic process / trypsin / digestion / response to nutrient / serine-type endopeptidase activity / calcium ion binding / proteolysis / : / extracellular region Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.8 Å X-RAY DIFFRACTION / Resolution: 2.8 Å | ||||||
 Authors Authors | Stroud, R.M. / Finer-Moore, J. | ||||||
 Citation Citation | Journal: Science / Year: 1987 Title: The three-dimensional structure of Asn102 mutant of trypsin: role of Asp102 in serine protease catalysis. Authors: Sprang, S. / Standing, T. / Fletterick, R.J. / Stroud, R.M. / Finer-Moore, J. / Xuong, N.H. / Hamlin, R. / Rutter, W.J. / Craik, C.S. #1: Journal: Science / Year: 1987Title: The Catalytic Role of the Active Site Aspartic Acid in Serine Proteases Authors: Craik, C.S. / Roczniak, S. / Largman, C. / Rutter, W.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2trm.cif.gz | 58.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2trm.ent.gz | 41.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2trm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tr/2trmftp://data.pdbj.org/pub/pdb/validation_reports/tr/2trm | HTTPS FTP |
|---|
-Related structure data




















-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23813.854 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #3: Chemical | ChemComp-BEN / 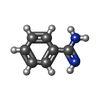  Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 122 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 122 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | THE CATALYTIC SITE, DIFFERS FROM THE CATALYTIC SITE OF NATIVE TRYPSIN BY REPLACEMENT OF ASP 102 ...THE CATALYTIC SITE, DIFFERS FROM THE CATALYTIC SITE OF NATIVE TRYPSIN BY REPLACEMEN |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.37 Å3/Da / Density % sol: 63.45 % |
|---|---|
| Crystal grow | Details: THE CRYSTALS WERE GROWN AT PH 8 WHICH IS WITHIN THE PH RANGE (7 - 9) WHERE NATIVE TRYPSIN IS OPTIMALLY ACTIVE. |
| Crystal grow | *PLUS Method: vapor diffusion |
-Data collection
| Reflection | *PLUS Highest resolution: 2.8 Å / Num. obs: 4500 / Num. measured all: 5000 |
|---|
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.8→7 Å / Rfactor obs: 0.157 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→7 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.8 Å / Lowest resolution: 7 Å / Rfactor obs: 0.157 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: p_angle_d |
