 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2rk1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | DHFR R67 Complexed with NADP and dihydrofolate | ||||||
 Components Components | Dihydrofolate reductase type 2 | ||||||
 Keywords Keywords | OXIDOREDUCTASE / NADP / dihydrofolate / asymmetric ligand binding / Antibiotic resistance / Methotrexate resistance / One-carbon metabolism / Plasmid / Trimethoprim resistance | ||||||
| Function / homology |  Function and homology information Function and homology informationresponse to methotrexate / dihydrofolate reductase / dihydrofolate reductase activity / tetrahydrofolate biosynthetic process / one-carbon metabolic process / response to xenobiotic stimulus / response to antibiotic Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.26 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.26 Å | ||||||
 Authors Authors | Krahn, J.M. / London, R.E. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2007 Title: Crystal structure of a type II dihydrofolate reductase catalytic ternary complex. Authors: Krahn, J.M. / Jackson, M.R. / DeRose, E.F. / Howell, E.E. / London, R.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2rk1.cif.gz | 61.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2rk1.ent.gz | 45.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2rk1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rk/2rk1ftp://data.pdbj.org/pub/pdb/validation_reports/rk/2rk1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2rh2SC  2rk2C S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj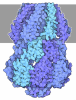




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a tetramer generated from the monomer in the asymmetric unit with the symmetry operations: x, y, z; y, x, -z; -x, -y, z; -y, -x, -z; The NADP and dihydrofolate ligands should be included only for the identity operator, and assigned full occupancy. Water molecules that clash with NADP or dihydrofolate should then be removed to obtain the set of waters that are valid for the tetramer. Tyr69 and Gln67 are also asymmetric, but the correlation of their alternate conformations to the asymmetric ligand binding has not been conclusively determined. |
-Components
| #1: Protein | Mass: 6732.528 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Strain: TMP-RESISTANT, CONTAINING R67 DHFR OVERPRODUCING PLASMID PLZ1 References: UniProt: P00383, dihydrofolate reductase | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | 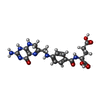  Mass: 443.413 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H21N7O6 Mass: 443.413 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H21N7O6#3: Chemical | ChemComp-NAP / |   Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3 Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3#4: Chemical |   Mass: 118.174 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 109 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 109 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 44.79 % |
|---|---|
| Crystal grow | Temperature: 277 K / pH: 8 Details: 50% 2-methyl-2,4-pentanediol (MPD), 100 mM Tris, pH 8.0, VAPOR DIFFUSION, HANGING DROP, temperature 277K, pH 8.00 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.5418 |
| Detector | Type: RIGAKU SATURN 92 / Detector: CCD / Date: Mar 17, 2005 / Details: VARIMAX HF, CONFOCAL MIRRORS |
| Radiation | Monochromator: CONFOCAL MIRRORS (VARIMAX HF) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.26→50 Å / Num. obs: 15838 / % possible obs: 99.6 % / Observed criterion σ(I): -3 / Redundancy: 12.4 % / Rmerge(I) obs: 0.072 / Net I/σ(I): 14.1 |
| Reflection shell | Resolution: 1.26→1.29 Å / Redundancy: 5.4 % / Rmerge(I) obs: 0.499 / Mean I/σ(I) obs: 3 / % possible all: 96.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2RH2 Resolution: 1.26→26.22 Å / Cor.coef. Fo:Fc: 0.974 / Cor.coef. Fo:Fc free: 0.967 / SU B: 1.057 / SU ML: 0.021 / Isotropic thermal model: Anisotropic / Cross valid method: THROUGHOUT / ESU R: 0.045 / ESU R Free: 0.042 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.403 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.26→26.22 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.26→1.293 Å / Total num. of bins used: 20
|
