 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2pl7: Orhorhombic crystal structure of hydrophobin HFBII in the presenc... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2pl7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Orhorhombic crystal structure of hydrophobin HFBII in the presence of a detergent | ||||||
 Components Components | Hydrophobin-2 | ||||||
 Keywords Keywords | SURFACE ACTIVE PROTEIN / hydrophobin / amphiphile / protein surfactant | ||||||
| Function / homology |  Function and homology information Function and homology informationconidium formation / spore wall / sporulation resulting in formation of a cellular spore / extracellular region Similarity search - Function | ||||||
| Biological species |  Hypocrea jecorina (fungus) Hypocrea jecorina (fungus) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1 Å | ||||||
 Authors Authors | Kallio, J.M. / Rouvinen, J.P. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2007 Title: Crystal Structures of Hydrophobin HFBII in the Presence of Detergent Implicate the Formation of Fibrils and Monolayer Films. Authors: Kallio, J.M. / Linder, M.B. / Rouvinen, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2pl7.cif.gz | 67.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2pl7.ent.gz | 49.4 KB | Display | PDB format |
| PDBx/mmJSON format | 2pl7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pl/2pl7ftp://data.pdbj.org/pub/pdb/validation_reports/pl/2pl7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2pl6C  1r2mS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 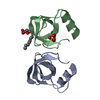
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 7201.474 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) Hypocrea jecorina (fungus) / References: UniProt: P79073#2: Chemical | ChemComp-SO4 / |   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#3: Sugar | ChemComp-HTG / | 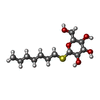  Type: D-saccharide / Mass: 294.408 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H26O5S / Comment: detergent*YM Type: D-saccharide / Mass: 294.408 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H26O5S / Comment: detergent*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 149 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 149 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.92 Å3/Da / Density % sol: 31.918 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.91 Details: 0.1 M lithium sulphate, 20% polyethylene glycol (MW2000), 0.1M Tris (pH 8.5), pH 6.91, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X12 / Wavelength: 0.9 Å / Beamline: X12 / Wavelength: 0.9 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Oct 3, 2006 |
| Radiation | Monochromator: Double crystal Si[111], horizontally focussing Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 1→20 Å / Num. all: 109088 / Num. obs: 107245 / % possible obs: 98.3 % / Observed criterion σ(I): -3 / Redundancy: 3.4 % / Biso Wilson estimate: 8.243 Å2 / Rmerge(I) obs: 0.101 / Net I/σ(I): 8.82 |
| Reflection shell | Resolution: 1→1.2 Å / Redundancy: 2.35 % / Rmerge(I) obs: 0.242 / Mean I/σ(I) obs: 3.9 / Num. measured obs: 104770 / Num. unique all: 44537 / % possible all: 96.9 |
-Phasing
| Phasing MR |
|
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1R2M Resolution: 1→20 Å / Num. parameters: 10287 / Num. restraintsaints: 12647 / Isotropic thermal model: anisotropic / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER
| |||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 10.2 Å2 | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 8 / Occupancy sum hydrogen: 948 / Occupancy sum non hydrogen: 1073.52 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1→20 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
