Entry Database : PDB / ID : 2omhTitle Structure of human insulin cocrystallized with ARG-12 peptide in presence of urea Insulin A chain Insulin B chain Keywords / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 1.36 Å Authors Norrman, M. / Schluckebier, G. Journal : EUR.J.PHARM.SCI. / Year : 2007Title : Structural characterization of insulin NPH formulations.Authors : Norrman, M. / Hubalek, F. / Schluckebier, G. History Deposition Jan 22, 2007 Deposition site / Processing site Revision 1.0 Mar 27, 2007 Provider / Type Revision 1.1 May 1, 2008 Group Revision 1.2 Jul 13, 2011 Group / Derived calculations / Version format complianceRevision 1.3 Mar 7, 2018 Group / Category / Item Revision 1.4 Dec 27, 2023 Group / Database references / Derived calculationsCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_struct_conn_angle / struct_conn / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_asym_id / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_leaving_atom_flag / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id Revision 1.5 Apr 3, 2024 Group / Category
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
















 PDBj
PDBj

















 Assembly
Assembly







 Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
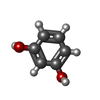
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 110.111 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H6O2
Mass: 110.111 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H6O2 Mass: 60.055 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: CH4N2O
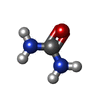
Mass: 60.055 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: CH4N2O Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Type: L-peptide NH3 amino terminus / Mass: 45.041 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH3NO
Type: L-peptide NH3 amino terminus / Mass: 45.041 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH3NO Sample preparation
Sample preparation / Beamline: I911-2 / Wavelength: 1 Å
/ Beamline: I911-2 / Wavelength: 1 Å Processing
Processing