SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED.
Resolution: 1.45→25.38 Å / Cor.coef. Fo:Fc: 0.968 / Cor.coef. Fo:Fc free: 0.965 / SU B: 1.93 / SU ML: 0.043 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.062 / ESU R Free: 0.06 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.191
2355
5 %
RANDOM
Rwork
0.178
-
-
-
obs
0.179
45070
98.9 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information MYCOBACTERIUM SMEGMATIS (bacteria)
MYCOBACTERIUM SMEGMATIS (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj



 Assembly
Assembly

 Mass: 54.938 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mn
Mass: 54.938 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mn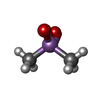
 Mass: 136.989 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6AsO2
Mass: 136.989 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6AsO2
 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 18.015 Da / Num. of mol.: 240 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 240 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID14-2 / Wavelength: 0.933
/ Beamline: ID14-2 / Wavelength: 0.933  Processing
Processing