 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2f95: M intermediate structure of sensory rhodopsin II/transducer compl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2f95 | ||||||
|---|---|---|---|---|---|---|---|
| Title | M intermediate structure of sensory rhodopsin II/transducer complex in combination with the ground state structure | ||||||
 Components Components |
| ||||||
 Keywords Keywords | MEMBRANE PROTEIN / MEMBRANE PROTEIN COMPLEX / SIGNAL TRANSDUCTION / PHOTOCYCLE STATE | ||||||
| Function / homology |  Function and homology information Function and homology informationphotoreceptor activity / phototransduction / monoatomic ion channel activity / chemotaxis / transmembrane signaling receptor activity / signal transduction / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  Natronomonas pharaonis (archaea) Natronomonas pharaonis (archaea) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Moukhametzianov, R.I. / Klare, J.P. / Efremov, R.G. / Baecken, C. / Goeppner, A. / Labahn, J. / Engelhard, M. / Bueldt, G. / Gordeliy, V.I. | ||||||
 Citation Citation | Journal: Nature / Year: 2006 Title: Development of the signal in sensory rhodopsin and its transfer to the cognate transducer. Authors: Moukhametzianov, R. / Klare, J.P. / Efremov, R. / Baeken, C. / Goppner, A. / Labahn, J. / Engelhard, M. / Buldt, G. / Gordeliy, V.I. #1: Journal: Nature / Year: 2002Title: Molecular basis of transmembrane signalling by sensory rhodopsin II - transducer complex Authors: Gordeliy, V.I. / Labahn, J. / Moukhametzianov, R.I. / Efremov, R.G. / Granzin, J. / Schlesinger, R. / Bueldt, G. / Savapol, T. / Scheidig, A.J. / Klare, J.P. / Engelhard, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2f95.cif.gz | 116.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2f95.ent.gz | 89.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2f95.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/f9/2f95ftp://data.pdbj.org/pub/pdb/validation_reports/f9/2f95 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2f93C  1h2sS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Number of models | 2 | ||||||||
| Details | Biological unit is thought to consist of one ground state complex of sensory rhodopsin II/transducer and one M state complex which is positioned relative to ground state by application of two fold operator: 1-x, -y, z |
-Components
| #1: Protein | Mass: 26534.941 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Natronomonas pharaonis (archaea) / Gene: sop2, sopII / Plasmid: PET27BMOD / Species (production host): Escherichia coli / Production host:  |
|---|---|
| #2: Protein | Mass: 17224.270 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Natronomonas pharaonis (archaea) / Gene: htr2, htrII / Plasmid: PET27BMOD / Species (production host): Escherichia coli / Production host: |
| #3: Sugar | ChemComp-BOG / 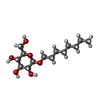  Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 1 Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C14H28O6 / Comment: detergent*YM |
| #4: Chemical | ChemComp-RET /   Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.8 Å3/Da / Density % sol: 31.6 % |
|---|---|
| Crystal grow | Temperature: 296 K / pH: 5.8 Details: 150 MM NACL, 25 MM NAKPI 5.1 0.8% B-OCTYLGLUCOSID , MONOVACCENIN (CUBIC PHASE) PRECIPITATED BY 1 M NA/KPI 5.8 ,in cubic lipidic phase, temperature 296K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-1 / Wavelength: 0.934 Å / Beamline: ID14-1 / Wavelength: 0.934 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Sep 20, 2004 |
| Radiation | Monochromator: diamond / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.934 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→22.01 Å / Num. all: 16493 / Num. obs: 16147 / % possible obs: 97.9 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 1.8 / Redundancy: 3.9 % / Biso Wilson estimate: 25.2 Å2 / Rmerge(I) obs: 0.085 |
| Reflection shell | Resolution: 2.2→2.26 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.414 / % possible all: 99.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB Entry: 1H2S Resolution: 2.2→22.01 Å / Rfactor Rfree error: 0.009 / Data cutoff high absF: 2058714.26 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: Engh & Huber Details: MODEL USED FOR REFINEMENT CONSISTS OF GROUND STATE COMPLEX (ALTERNATIVE CONFORMATION INDICATOR A AT POSITION 17) AND M STATE COMPLEX (ALTERNATIVE CONFORMATION INDICATOR B AT POSITION 17) ...Details: MODEL USED FOR REFINEMENT CONSISTS OF GROUND STATE COMPLEX (ALTERNATIVE CONFORMATION INDICATOR A AT POSITION 17) AND M STATE COMPLEX (ALTERNATIVE CONFORMATION INDICATOR B AT POSITION 17) WITH CORRESPONDING OCCUPANCIES. GROUND STATE MODEL WAS FIRST REFINED AGAINST THE GROUND STATE DATA AND THEN THE M STATE MODEL WAS REFINED WITH GROUND STATE MODEL FIXED AGAINST THE ILLUMINATED CRYSTAL DATA
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 59.764 Å2 / ksol: 0.347059 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→22.01 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.28 Å / Rfactor Rfree error: 0.033 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
