 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2eat | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the SR CA2+-ATPASE with bound CPA and TG | ||||||
 Components Components | Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 | ||||||
 Keywords Keywords | HYDROLASE / MEMBRANE PROTEIN / P-TYPE ATPASE / HAD FOLD / Ca2+ / ion pump | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of calcium ion import into sarcoplasmic reticulum / positive regulation of ATPase-coupled calcium transmembrane transporter activity / positive regulation of fast-twitch skeletal muscle fiber contraction / H zone / calcium ion import into sarcoplasmic reticulum / negative regulation of striated muscle contraction / regulation of striated muscle contraction / P-type Ca2+ transporter / P-type calcium transporter activity / positive regulation of cardiac muscle cell contraction ...positive regulation of calcium ion import into sarcoplasmic reticulum / positive regulation of ATPase-coupled calcium transmembrane transporter activity / positive regulation of fast-twitch skeletal muscle fiber contraction / H zone / calcium ion import into sarcoplasmic reticulum / negative regulation of striated muscle contraction / regulation of striated muscle contraction / P-type Ca2+ transporter / P-type calcium transporter activity / positive regulation of cardiac muscle cell contraction / I band / endoplasmic reticulum-Golgi intermediate compartment / sarcoplasmic reticulum membrane / sarcoplasmic reticulum / calcium ion transport / calcium ion binding / endoplasmic reticulum membrane / perinuclear region of cytoplasm / endoplasmic reticulum / ATP hydrolysis activity / ATP binding / membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | ||||||
 Authors Authors | Takahashi, M. / Kondou, Y. / Toyoshima, C. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.Usa / Year: 2007 Title: Interdomain communication in calcium pump as revealed in the crystal structures with transmembrane inhibitors Authors: Takahashi, M. / Kondou, Y. / Toyoshima, C. #1: Journal: Nature / Year: 2000Title: Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution Authors: Toyoshima, C. / Nakasako, M. / Nomura, H. / Ogawa, H. #2: Journal: Nature / Year: 2002Title: Structural changes in the calcium pump accompanying the dissociation of calcium Authors: Toyoshima, C. / Nomura, H. #3: Journal: Nature / Year: 2004Title: Crystal structure of the calcium pump with a bound ATP analogue Authors: Toyoshima, C. / Mizutani, T. #4: Journal: Nature / Year: 2004Title: Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues Authors: Toyoshima, C. / Nomura, H. / Tsuda, T. #5: Journal: Proc.Natl.Acad.Sci.Usa / Year: 2005Title: Structural role of countertransport revealed in Ca(2+) pump crystal structure in the absence of Ca(2+) Authors: Obara, K. / Miyashita, N. / Xu, C. / Toyoshima, I. / Sugita, Y. / Inesi, G. / Toyoshima, C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2eat.cif.gz | 204.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2eat.ent.gz | 161.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2eat.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ea/2eatftp://data.pdbj.org/pub/pdb/validation_reports/ea/2eat | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2earC  2eauC  4yclC  1iwoS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |





















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 109628.617 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Chemical | ChemComp-TG1 / 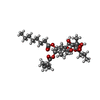  Mass: 650.754 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H50O12 Mass: 650.754 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H50O12 |
| #3: Chemical | ChemComp-CZA / (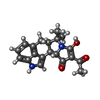  Mass: 336.384 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H20N2O3 Mass: 336.384 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H20N2O3 |
| Has protein modification | Y |
| Sequence details | THE C-TERMINAL RESIDUES IN UNP ENTRY P04191 ARE FROM 994 TO 1001, DPEDERRK. IN ISOFORM SERCA1A, ...THE C-TERMINAL RESIDUES IN UNP ENTRY P04191 ARE FROM 994 TO 1001, DPEDERRK. IN ISOFORM SERCA1A, THERE IS ONLY ONE C-TERMINAL RESIDUE 994 GLY. |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.24 Å3/Da / Density % sol: 70.98 % |
|---|---|
| Crystal grow | Temperature: 283 K / Method: microdialysis / pH: 6.1 / Details: PEG 400, pH 6.1, MICRODIALYSIS, temperature 283K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL41XU / Wavelength: 0.8 Å / Beamline: BL41XU / Wavelength: 0.8 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Dec 12, 2005 |
| Radiation | Monochromator: ROTATED-INCLINED DOUBLE-CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→20 Å / Num. all: 40669 / Num. obs: 40344 / % possible obs: 99.2 % / Observed criterion σ(F): 6.13 / Observed criterion σ(I): -2 / Redundancy: 7 % / Rmerge(I) obs: 0.048 / Net I/σ(I): 33.6 |
| Reflection shell | Resolution: 2.9→2.97 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.361 / Mean I/σ(I) obs: 2 / % possible all: 92.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1IWO Resolution: 2.9→15 Å / Cor.coef. Fo:Fc: 0.932 / Cor.coef. Fo:Fc free: 0.894 / SU B: 33.458 / SU ML: 0.296 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.746 / ESU R Free: 0.388 / Stereochemistry target values: maximum likelihood
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 77.42 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.9→2.972 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
