Entry Database : PDB / ID : 2bjsTitle Isopenicillin N synthase C-terminal truncation mutant ISOPENICILLIN N SYNTHETASE Keywords / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Emericella nidulans (mold)Method / / / Resolution : 1.3 Å Authors McNeill, L.A. / Sami, M. / Clifton, I.J. / Burzlaff, N.I. Journal : Chemistry / Year : 2017Title : Terminally Truncated Isopenicillin N Synthase Generates a Dithioester Product: Evidence for a Thioaldehyde Intermediate during Catalysis and a New Mode of Reaction for Non-Heme Iron Oxidases.Authors : McNeill, L.A. / Brown, T.J.N. / Sami, M. / Clifton, I.J. / Burzlaff, N.I. / Claridge, T.D.W. / Adlington, R.M. / Baldwin, J.E. / Rutledge, P.J. / Schofield, C.J. History Deposition Feb 7, 2005 Deposition site / Processing site Revision 1.0 Mar 9, 2006 Provider / Type Revision 1.1 Jul 13, 2011 Group / Version format complianceRevision 1.2 Dec 6, 2017 Group / Category / citation_authorItem _citation.country / _citation.journal_abbrev ... _citation.country / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year Revision 1.3 Feb 21, 2018 Group / Category / Item Revision 1.4 Dec 13, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id Revision 1.5 May 8, 2024 Group / Category Item / _entity_src_gen.pdbx_gene_src_ncbi_taxonomy_id / _entity_src_gen.pdbx_gene_src_scientific_name
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads





































 PDBj
PDBj
 Assembly
Assembly


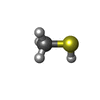


 Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe
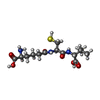
Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe Mass: 363.430 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H25N3O6S
Mass: 363.430 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H25N3O6S Mass: 48.107 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH4S
Mass: 48.107 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH4S Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Sample preparation
Sample preparation / Beamline: ID14-3 / Wavelength: 0.93487
/ Beamline: ID14-3 / Wavelength: 0.93487  Processing
Processing