 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2b1i: crystal structures of transition state analogue inhibitors of ino... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2b1i | ||||||
|---|---|---|---|---|---|---|---|
| Title | crystal structures of transition state analogue inhibitors of inosine monophosphate cyclohydrolase | ||||||
 Components Components | Bifunctional purine biosynthesis protein PURH | ||||||
 Keywords Keywords | TRANSFERASE / HYDROLASE / ATIC / IMPCH / structure-base / transition state analogue / inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationDe novo synthesis of IMP / phosphoribosylaminoimidazolecarboxamide formyltransferase / phosphoribosylaminoimidazolecarboxamide formyltransferase activity / IMP cyclohydrolase / IMP cyclohydrolase activity / 'de novo' IMP biosynthetic process / protein homodimerization activity / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.02 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.02 Å | ||||||
 Authors Authors | Xu, L. / Chong, Y. / Hwang, I. / D'Onofrio, A. / Amore, K. / Beardsley, G.P. / Li, C. / Olson, A.J. / Boger, D.L. / Wilson, I.A. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2007 Title: Structure-based Design, Synthesis, Evaluation, and Crystal Structures of Transition State Analogue Inhibitors of Inosine Monophosphate Cyclohydrolase. Authors: Xu, L. / Chong, Y. / Hwang, I. / D'Onofrio, A. / Amore, K. / Beardsley, G.P. / Li, C. / Olson, A.J. / Boger, D.L. / Wilson, I.A. #1: Journal: J.Biol.Chem. / Year: 2004Title: Crystal structure of avian AICAR transformylase in complex with a novel non-folate inhibitor identified by virtual ligand screening Authors: Xu, L. / Li, C. / Olson, A.J. / Wilson, I.A. #2: Journal: J.Med.Chem. / Year: 2004Title: Successful virtual screening for human AICAR transformylase inhibitors against NCI diversity set using AutoDock. Authors: Li, C. / Xu, L. / Wolan, D.W. / Wilson, I.A. / Olson, A.J. #3: Journal: Nat.Struct.Biol. / Year: 2001Title: Crystal structure of a bifunctional transformylase and cyclohydrolase enzyme in purine biosynthesis Authors: Greasley, S.E. / Horton, P. / Ramcharan, J. / Beardsley, G.P. / Benkovic, S.J. / Wilson, I.A. #4: Journal: Biochemistry / Year: 2002Title: Structural insights into the avian AICAR transformylase mechanism Authors: Wolan, D.W. / Greasley, S.E. / Beardsley, G.P. / Wilson, I.A. #5: Journal: Biochemistry / Year: 2003Title: Structure of avian AICAR transformylase with a multisubstrate adduct inhibitor beta-DADF identifies the folate binding site Authors: Wolan, D.W. / Greasley, S.E. / Wall, M.J. / Benkovic, S.J. / Wilson, I.A. #6: Journal: Biochemistry / Year: 2004Title: Structural insights into the human and avian IMP cyclohydrolase mechanism via crystal structures with the bound XMP Inhibitor Authors: Wolan, D.W. / Cheong, C.-G. / Greasley, S.E. / Wilson, I.A. #7: Journal: J.Biol.Chem. / Year: 2004Title: Crystal structures of human bifunctional enzyme aminoimidazole-4-carboxamide ribonucleotide transformylase/IMP cyclohydrolase in complex with potent sulfonyl-containing antifolates Authors: Cheong, C.G. / Wolan, D.W. / Greasley, S.E. / Horton, P.A. / Beardsley, G.P. / Wilson, I.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2b1i.cif.gz | 464.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2b1i.ent.gz | 382 KB | Display | PDB format |
| PDBx/mmJSON format | 2b1i.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 2b1i_validation.pdf.gz | 1.7 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 2b1i_full_validation.pdf.gz | 1.7 MB | Display | |
| Data in XML | 2b1i_validation.xml.gz | 47.9 KB | Display | |
| Data in CIF | 2b1i_validation.cif.gz | 66.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b1/2b1iftp://data.pdbj.org/pub/pdb/validation_reports/b1/2b1i | HTTPS FTP |
-Related structure data
| Related structure data |  2b1gC  2iu0C  2iu3C  1g8mS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological unit is a dimer. There is 1 biological unit in the asymmetric unit (chains A & B) |
-Components
| #1: Protein | Mass: 64497.703 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: Includes: Phosphoribosylaminoimidazolecarboxamide formyltransferase (AICAR transformylase), IMP cyclohydrolase (Inosinicase, IMP synthetase, ATIC) Source: (gene. exp.)  References: UniProt: P31335, IMP cyclohydrolase, phosphoribosylaminoimidazolecarboxamide formyltransferase #2: Chemical |   Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K#3: Chemical | ChemComp-93A / [ 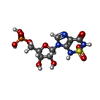  Mass: 400.259 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H13N4O10PS Mass: 400.259 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H13N4O10PS#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 45.7 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / pH: 7.2 Details: PEG 8000, imidazole, DTT, pH 7.2, VAPOR DIFFUSION, SITTING DROP, temperature 295K |
-Data collection
| Diffraction | Mean temperature: 93 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.2.1 / Wavelength: 0.99997 Å / Beamline: 8.2.1 / Wavelength: 0.99997 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Nov 11, 2004 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: double crystals / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.99997 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Number: 75375 / Rmerge(I) obs: 0.065 / Χ2: 2.621 / D res high: 2.02 Å / D res low: 50 Å / % possible obs: 97.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.02→50 Å / Num. obs: 75375 / % possible obs: 97.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.5 % / Biso Wilson estimate: 31.2 Å2 / Rsym value: 0.065 / Χ2: 2.621 / Net I/σ(I): 24.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Resolution: 2.02→2.09 Å / % possible obs: 91.5 % / Redundancy: 2 % / Mean I/σ(I) obs: 2.8 / Num. measured obs: 7071 / Num. unique all: 7071 / Rsym value: 0.275 / Χ2: 1.105 / % possible all: 91.5 |
-Phasing
| Phasing MR | Rfactor: 43.6 / Cor.coef. Fo:Fc: 49.2 / Cor.coef. Io to Ic: 50.3
|
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1G8M Resolution: 2.02→38.07 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.924 / SU B: 10.998 / SU ML: 0.152 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.231 / ESU R Free: 0.194 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 41.161 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.02→38.07 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.018→2.071 Å / Total num. of bins used: 20
|
