 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1m9n: CRYSTAL STRUCTURE OF THE HOMODIMERIC BIFUNCTIONAL TRANSFORMYLASE ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1m9n | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE HOMODIMERIC BIFUNCTIONAL TRANSFORMYLASE AND CYCLOHYDROLASE ENZYME AVIAN ATIC IN COMPLEX WITH AICAR AND XMP AT 1.93 ANGSTROMS. | |||||||||
 Components Components | AICAR TRANSFORMYLASE-IMP CYCLOHYDROLASE | |||||||||
 Keywords Keywords | transferase / hydrolase / HOMODIMER / 2 FUNCTIONAL DOMAINS / IMPCH DOMAIN = ALPHA/BETA/ALPHA / AICAR TFASE = 2 ALPHA/BETA/ALPHA DOMAINS / 1 ALPHA + BETA DOMAIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationDe novo synthesis of IMP / phosphoribosylaminoimidazolecarboxamide formyltransferase / phosphoribosylaminoimidazolecarboxamide formyltransferase activity / IMP cyclohydrolase / IMP cyclohydrolase activity / 'de novo' IMP biosynthetic process / protein homodimerization activity / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.93 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.93 Å | |||||||||
 Authors Authors | Wolan, D.W. / Greasly, S.E. / Beardsley, G.P. / Wilson, I.A. | |||||||||
 Citation Citation | Journal: Biochemistry / Year: 2002 Title: Structural Insights into the Avian AICAR Transformylase Mechanism. Authors: Wolan, D.W. / Greasly, S.E. / Beardsley, G.P. / Wilson, I.A. | |||||||||
| History |
| |||||||||
| Remark 12 | OTHER REFINEMENT REMARKS Reported B value is prior to incorporation of tensors with TLSANL. Tensors ...OTHER REFINEMENT REMARKS Reported B value is prior to incorporation of tensors with TLSANL. Tensors have been added to the B value for the publication. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1m9n.cif.gz | 248.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1m9n.ent.gz | 195.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1m9n.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/m9/1m9nftp://data.pdbj.org/pub/pdb/validation_reports/m9/1m9n | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1g8mS S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | Both monomers of the biologically significant avian ATIC homodimer are located within the asymmetric unit. |
-Components
| #1: Protein | Mass: 66669.086 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  References: UniProt: P31335, phosphoribosylaminoimidazolecarboxamide formyltransferase, IMP cyclohydrolase #2: Chemical |   Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K#3: Chemical |   Mass: 338.211 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H15N4O8P Mass: 338.211 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H15N4O8P#4: Chemical | 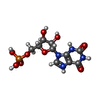  Mass: 365.213 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H14N4O9P Mass: 365.213 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H14N4O9P#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 511 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 511 / Source method: isolated from a natural source / Formula: H2OHas protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.37 Å3/Da / Density % sol: 48.16 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / pH: 7.2 Details: PEG 8000, imidazole, DTT, pH 7.2, VAPOR DIFFUSION, SITTING DROP, temperature 295K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-BM-C / Wavelength: 1 Å / Beamline: 14-BM-C / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Sep 16, 1999 Details: bent conical Si-mirror (Rh coating); bent cylindrical Ge(111) monochromator |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.93→30 Å / Num. all: 92862 / Num. obs: 91184 / % possible obs: 98.2 % / Observed criterion σ(I): -3 / Redundancy: 3.9 % / Biso Wilson estimate: 33.5 Å2 / Rsym value: 0.044 / Net I/σ(I): 25.3 |
| Reflection shell | Resolution: 1.93→1.96 Å / Redundancy: 3.4 % / Mean I/σ(I) obs: 2.4 / Num. unique all: 4619 / Rsym value: 0.419 / % possible all: 99.6 |
| Reflection | *PLUS Lowest resolution: 30 Å / Rmerge(I) obs: 0.044 |
| Reflection shell | *PLUS % possible obs: 99.6 % / Num. unique obs: 4619 / Rmerge(I) obs: 0.419 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1G8M Resolution: 1.93→29.24 Å / Cor.coef. Fo:Fc: 0.953 / Cor.coef. Fo:Fc free: 0.934 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.8 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.949 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.93→29.24 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.933→1.983 Å / Total num. of bins used: 20 /
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 29.2 Å / % reflection Rfree: 10 % / Rfactor Rfree: 0.244 / Rfactor Rwork: 0.206 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.93 Å / Lowest resolution: 1.98 Å |
