 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-212l: PROTEIN STRUCTURE PLASTICITY EXEMPLIFIED BY INSERTION AND DELETIO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 212l | ||||||
|---|---|---|---|---|---|---|---|
| Title | PROTEIN STRUCTURE PLASTICITY EXEMPLIFIED BY INSERTION AND DELETION MUTANTS IN T4 LYSOZYME | ||||||
 Components Components | T4 LYSOZYME | ||||||
 Keywords Keywords | HYDROLASE (O-GLYCOSYL) / GLYCOSIDASE / BACTERIOLYTIC ENZYME | ||||||
| Function / homology |  Function and homology information Function and homology informationviral release from host cell by cytolysis / peptidoglycan catabolic process / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / host cell cytoplasm / defense response to bacterium Similarity search - Function | ||||||
| Biological species |  Enterobacteria phage T4 (virus) Enterobacteria phage T4 (virus) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.76 Å X-RAY DIFFRACTION / Resolution: 1.76 Å | ||||||
 Authors Authors | Vetter, I.R. / Baase, W.A. / Heinz, D.W. / Xiong, J.-P. / Snow, S. / Matthews, B.W. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 1996 Title: Protein structural plasticity exemplified by insertion and deletion mutants in T4 lysozyme. Authors: Vetter, I.R. / Baase, W.A. / Heinz, D.W. / Xiong, J.P. / Snow, S. / Matthews, B.W. #1: Journal: Nature / Year: 1993Title: How Amino-Acid Insertions are Allowed in an Alpha-Helix of T4 Lysozyme Authors: Heinz, D.W. / Baase, W.A. / Dahlquist, F.W. / Matthews, B.W. #2: Journal: J.Mol.Biol. / Year: 1987Title: Structure of Bacteriophage T4 Lysozyme Refined at 1.7 A Resolution Authors: Weaver, L.H. / Matthews, B.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 212l.cif.gz | 48.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb212l.ent.gz | 34.6 KB | Display | PDB format |
| PDBx/mmJSON format | 212l.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/12/212lftp://data.pdbj.org/pub/pdb/validation_reports/12/212l | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  209lC  210lC  211lC  213lC  214lC  215lC  218lC  219lC C: citing same article ( |
|---|---|
| Similar structure data |






















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18912.678 Da / Num. of mol.: 1 / Mutation: C54T, C97A, INS(L164-AAAA) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Enterobacteria phage T4 (virus) / Genus: T4-like viruses / Species: Enterobacteria phage T4 sensu lato / Gene: T4 LYSOZYME GENE / Plasmid: M13 / Gene (production host): T4 LYSOZYME GENE / Production host:  |
|---|---|
| #2: Chemical | ChemComp-HED / 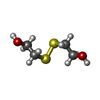  Mass: 154.251 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O2S2 Mass: 154.251 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O2S2 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 155 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 155 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.83 Å3/Da / Density % sol: 48.3 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Details: MUTANT CRYSTALLIZED FROM PEG IN CONTRAST TO L164AAAA WHICH WAS CRYSTALLIZED FROM PHOSPHATE. | ||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.5 / Method: unknown | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction source | Wavelength: 1.5418 |
|---|---|
| Detector | Type: XUONG-HAMLIN MULTIWIRE / Detector: AREA DETECTOR / Date: Nov 2, 1994 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.76→35.65 Å / Num. obs: 20392 / % possible obs: 95.3 % / Observed criterion σ(I): 0 / Redundancy: 2.4 % / Rmerge(I) obs: 0.0622 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.76→35.65 Å / σ(F): 0 Details: RESIDUES 162 - 168 IN THIS MUTANT LYSOZYME ARE EXTREMELY MOBILE. THUS THE COORDINATES FOR THESE RESIDUES ARE VERY UNRELIABLE. THIS MUTANT WAS CRYSTALLIZED FROM PEG, AS COMPARED TO ENTRY 219L ...Details: RESIDUES 162 - 168 IN THIS MUTANT LYSOZYME ARE EXTREMELY MOBILE. THUS THE COORDINATES FOR THESE RESIDUES ARE VERY UNRELIABLE. THIS MUTANT WAS CRYSTALLIZED FROM PEG, AS COMPARED TO ENTRY 219L WHICH IS THE SAME MUTANT, BUT CRYSTALLIZED FROM PHOSPHATE.
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.76→35.65 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: TNT / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.156 | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: t_plane_restr / Dev ideal: 0.019 / Weight: 3 |
