 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1zm2: Structure of ADP-ribosylated eEF2 in complex with catalytic fragm... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1zm2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of ADP-ribosylated eEF2 in complex with catalytic fragment of ETA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | BIOSYNTHETIC PROTEIN/TRANSFERASE / elongation factor / toxin / ADP-ribosylation / BIOSYNTHETIC PROTEIN-TRANSFERASE COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationsymbiont-mediated suppression of host translation elongation / NAD+-diphthamide ADP-ribosyltransferase / NAD+-diphthamide ADP-ribosyltransferase activity / Peptide chain elongation / Synthesis of diphthamide-EEF2 / positive regulation of translational elongation / symbiont-mediated killing of host cell / Protein methylation / translational elongation / translation elongation factor activity ...symbiont-mediated suppression of host translation elongation / NAD+-diphthamide ADP-ribosyltransferase / NAD+-diphthamide ADP-ribosyltransferase activity / Peptide chain elongation / Synthesis of diphthamide-EEF2 / positive regulation of translational elongation / symbiont-mediated killing of host cell / Protein methylation / translational elongation / translation elongation factor activity / nucleotidyltransferase activity / Neutrophil degranulation / maintenance of translational fidelity / toxin activity / protein-folding chaperone binding / ribosome binding / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / rRNA binding / ribonucleoprotein complex / GTPase activity / GTP binding / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.07 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.07 Å | ||||||
 Authors Authors | Joergensen, R. / Merrill, A.R. / Yates, S.P. / Marquez, V.E. / Schwan, A.L. / Boesen, T. / Andersen, G.R. | ||||||
 Citation Citation | Journal: Nature / Year: 2005 Title: Exotoxin A-eEF2 complex structure indicates ADP ribosylation by ribosome mimicry. Authors: Joergensen, R. / Merrill, A.R. / Yates, S.P. / Marquez, V.E. / Schwan, A.L. / Boesen, T. / Andersen, G.R. | ||||||
| History |
| ||||||
| Remark 400 | COMPOUND RESIDUE 699 IS AN ADP-RIBOSYLATED DIPHTHAMIDE WHICH IS COMPOSED OF DDE WITH A LINK TO APR ...COMPOUND RESIDUE 699 IS AN ADP-RIBOSYLATED DIPHTHAMIDE WHICH IS COMPOSED OF DDE WITH A LINK TO APR LIGANDS. THE COMPLETE ADP-RIBOSYLATED DIPHTHAMIDE CAN ONLY BE OBSERVED IN CHAIN E. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1zm2.cif.gz | 597.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1zm2.ent.gz | 482.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1zm2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zm/1zm2ftp://data.pdbj.org/pub/pdb/validation_reports/zm/1zm2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1zm3C  1zm4C  1zm9C  1aerS  1n0vS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 93549.320 Da / Num. of mol.: 3 / Fragment: eEF2 / Source method: isolated from a natural source / Source: (natural) #2: Protein | Mass: 22496.010 Da / Num. of mol.: 3 / Fragment: catalytic domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (bacteria) / Gene: eta / Production host: References: GenBank: 151216, UniProt: P11439*PLUS, Transferases; Glycosyltransferases; Pentosyltransferases #3: Chemical | ChemComp-APR / | 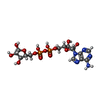  Mass: 559.316 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H23N5O14P2 Mass: 559.316 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H23N5O14P2 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.7 Å3/Da / Density % sol: 52 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7.2 Details: PEG 6000, MPD, HEPES, pH 7.2, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.952 Å / Beamline: 14.1 / Wavelength: 0.952 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Jan 13, 2005 |
| Radiation | Monochromator: Si-111 crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.952 Å / Relative weight: 1 |
| Reflection | Resolution: 3.07→40 Å / Num. all: 76524 / Num. obs: 75835 / % possible obs: 99.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.4 % / Rsym value: 0.142 / Net I/σ(I): 9.6 |
| Reflection shell | Resolution: 3.07→3.15 Å / % possible all: 91 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entries 1n0v and 1aer Resolution: 3.07→30 Å / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.07→30 Å
| ||||||||||||||||||||
| Refine LS restraints |
|
