 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1yfl: T4Dam in Complex with Sinefungin and 16-mer Oligonucleotide Showi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1yfl | ||||||
|---|---|---|---|---|---|---|---|
| Title | T4Dam in Complex with Sinefungin and 16-mer Oligonucleotide Showing Semi-specific and Specific Contact and Flipped Base | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE/DNA / T4DAM / METHYLTRANSFERASE / DNA / PROTEIN-DNA COMPLEX / BASE-FLIPPING / TRANSFERASE-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationsymbiont-mediated evasion of host restriction-modification system / site-specific DNA-methyltransferase (adenine-specific) / site-specific DNA-methyltransferase (adenine-specific) activity / DNA-methyltransferase activity / S-adenosyl-L-methionine binding / DNA restriction-modification system / mismatch repair / methylation / sequence-specific DNA binding / DNA replication / symbiont-mediated suppression of host innate immune response Similarity search - Function | ||||||
| Biological species |  Enterobacteria phage T4 (virus) Enterobacteria phage T4 (virus) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.09 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.09 Å | ||||||
 Authors Authors | Horton, J.R. / Liebert, K. / Hattman, S. / Jeltsch, A. / Cheng, X. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 2005 Title: Transition from Nonspecific to Specific DNA Interactions along the Substrate-Recognition Pathway of Dam Methyltransferase. Authors: Horton, J.R. / Liebert, K. / Hattman, S. / Jeltsch, A. / Cheng, X. #1: Journal: Nat.Struct.Mol.Biol. / Year: 2003Title: Structure of the bacteriophage T4 DNA adenine methyltransferase Authors: Yang, Z. / Horton, J.R. / Zhou, L. / Zhang, X.J. / Dong, A. / Zhang, X. / Schlagman, S.L. / Kossykh, V. / Cheng, X. | ||||||
| History |
| ||||||
| Remark 999 | SEQUENCE 1) Author states that Q139R, Y140F and Q209L reflect conflicts between deposited protein ...SEQUENCE 1) Author states that Q139R, Y140F and Q209L reflect conflicts between deposited protein sequence and translated deposited DNA-->protein sequence. From their electron density, it appears the translated DNA sequence is correct ; 2) It is possible residue 119 could be a Tyr rather than Asp based on some electron density evidence. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1yfl.cif.gz | 246 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1yfl.ent.gz | 193.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1yfl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yf/1yflftp://data.pdbj.org/pub/pdb/validation_reports/yf/1yfl | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1yf3C  1yfjC  1q0sS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj







































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 4898.192 Da / Num. of mol.: 4 / Source method: obtained synthetically / Details: Synthesized by New England Biolabs #2: Protein | Mass: 30461.898 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Enterobacteria phage T4 (virus) / Genus: T4-like viruses / Species: Enterobacteria phage T4 sensu lato / Gene: DAM / Plasmid: pJW2 / Production host:  References: UniProt: P04392, site-specific DNA-methyltransferase (adenine-specific) #3: Chemical | ChemComp-SFG / 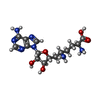  Mass: 381.387 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C15H23N7O5 Mass: 381.387 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C15H23N7O5#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.15 Å3/Da / Density % sol: 64.2 % | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, hanging drop / pH: 6.2 Details: PEG 6000, MES, ammonium acetate, CaCl2, glycerol, pH 6.2, VAPOR DIFFUSION, HANGING DROP, temperature 289K | ||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
|
-Data collection
| Diffraction | Mean temperature: 173 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-ID / Wavelength: 1 Å / Beamline: 22-ID / Wavelength: 1 Å |
| Detector | Type: MARRESEARCH / Detector: AREA DETECTOR / Date: Jun 6, 2004 |
| Radiation | Monochromator: Si 111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 3.09→30 Å / Num. obs: 33149 / % possible obs: 99.9 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 5.4 % / Biso Wilson estimate: 54.4 Å2 / Rsym value: 0.081 / Net I/σ(I): 18.7 |
| Reflection shell | Resolution: 3.09→3.2 Å / Redundancy: 5.4 % / Mean I/σ(I) obs: 5.8 / Num. unique all: 3302 / Rsym value: 0.314 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1Q0S Resolution: 3.09→29.4 Å / Isotropic thermal model: anisotropic / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: The crystal of T4Dam-sinefungin-16mer DNA can also be indexed in the space group P43, with the same cell dimensions but two protein molecules and two half DNA molecules in the asymmetric ...Details: The crystal of T4Dam-sinefungin-16mer DNA can also be indexed in the space group P43, with the same cell dimensions but two protein molecules and two half DNA molecules in the asymmetric unit. The DNA axis is in parallel with the crystallographic z-axis (a 4-fold screw axis). To have a complete DNA molecule, we indexed the diffraction data in a lower symmetry space group P21. The two protein molecules along the DNA axis were constrained during the refinement by non-crystallographic 4-fold screw symmetry.
| |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 57.7 Å2
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.09→29.4 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.09→3.2 Å / Rfactor Rfree error: 0.033
|
