 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1xz3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Complex of apoferritin with isoflurane | ||||||
 Components Components | Ferritin light chain | ||||||
 Keywords Keywords | METAL BINDING PROTEIN / 4-helix bundle | ||||||
| Function / homology |  Function and homology information Function and homology informationferritin complex / autolysosome / ferric iron binding / autophagosome / iron ion transport / ferrous iron binding / cytoplasmic vesicle / intracellular iron ion homeostasis / iron ion binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.75 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.75 Å | ||||||
 Authors Authors | Liu, R. / Loll, P.J. / Eckenhoff, R.G. | ||||||
 Citation Citation | Journal: Faseb J. / Year: 2005 Title: Structural basis for high-affinity volatile anesthetic binding in a natural 4-helix bundle protein. Authors: Liu, R. / Loll, P.J. / Eckenhoff, R.G. | ||||||
| History |
| ||||||
| Remark 999 | SEQUENCE Author states that residue 93 is indeed a LEU. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1xz3.cif.gz | 55.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1xz3.ent.gz | 40.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1xz3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xz/1xz3ftp://data.pdbj.org/pub/pdb/validation_reports/xz/1xz3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1xz1C  1gwgS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | x 24
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Details | Biological unit is 24-mer: x,y,z; -x,-y,z; -x,y,-z; x,-y,-z; z,x,y; z,-x,-y; -z,-x,y; -z,x,-y; y,z,x; -y,z,-x; y,-z,-x; -y,-z,x; y,x,-z; -y,-x,-z; y,-x,z; -y,x,z; x,z,-y; -x,z,y; -x,-z,-y; x,-z,y; z,y,-x; z,-y,x; -z,y,x; -z,-y,-x; |
-Components
| #1: Protein | Mass: 19872.428 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-CD /   Mass: 112.411 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: Cd Mass: 112.411 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: Cd#3: Chemical | ChemComp-ICF / | 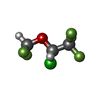  Mass: 184.492 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H2ClF5O Mass: 184.492 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H2ClF5O#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 190 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 190 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.15 Å3/Da / Density % sol: 60.9 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: cadmium sulfate, HEPES, sodium acetate, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X8C / Wavelength: 1.0722 Å / Beamline: X8C / Wavelength: 1.0722 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Mar 5, 2004 Details: parabolic collimating mirror upstream of monochromator |
| Radiation | Monochromator: parabolic collimating mirror upstream of monochromator Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0722 Å / Relative weight: 1 |
| Reflection | Resolution: 1.74→30 Å / Num. all: 25454 / Num. obs: 25454 / % possible obs: 95.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 10.4 % / Rmerge(I) obs: 0.064 / Net I/σ(I): 16.6 |
| Reflection shell | Resolution: 1.74→1.82 Å / Redundancy: 2.8 % / Rmerge(I) obs: 0.431 / Mean I/σ(I) obs: 2.2 / Num. unique all: 2551 / % possible all: 78.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 1GWG Resolution: 1.75→30 Å / Cor.coef. Fo:Fc: 0.956 / Cor.coef. Fo:Fc free: 0.94 / SU B: 2.185 / SU ML: 0.065 / TLS residual ADP flag: LIKELY RESIDUAL / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.097 / ESU R Free: 0.098 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 18.404 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.097 Å / Luzzati d res low obs: 0.098 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→30 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.75→1.795 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 28.5037 Å / Origin y: 9.6383 Å / Origin z: 39.6426 Å
|
