| Entry | Database: PDB / ID: 1w2c
|
|---|
| Title | Human Inositol (1,4,5) trisphosphate 3-kinase complexed with Mn2+/AMPPNP/Ins(1,4,5)P3 |
|---|
 Components Components | INOSITOL-TRISPHOSPHATE 3-KINASE A |
|---|
 Keywords Keywords | TRANSFERASE / INOSITOL PHOSPHATE KINASE / AMPPNP / IP3 |
|---|
| Function / homology |  Function and homology information Function and homology information
inositol-trisphosphate 3-kinase / inositol-1,4,5-trisphosphate 3-kinase activity / inositol hexakisphosphate kinase activity / inositol phosphate biosynthetic process / modification of postsynaptic actin cytoskeleton / postsynaptic actin cytoskeleton / calcium/calmodulin-dependent protein kinase activity / phosphatidylinositol phosphate biosynthetic process / Synthesis of IP3 and IP4 in the cytosol / cellular response to calcium ion ...inositol-trisphosphate 3-kinase / inositol-1,4,5-trisphosphate 3-kinase activity / inositol hexakisphosphate kinase activity / inositol phosphate biosynthetic process / modification of postsynaptic actin cytoskeleton / postsynaptic actin cytoskeleton / calcium/calmodulin-dependent protein kinase activity / phosphatidylinositol phosphate biosynthetic process / Synthesis of IP3 and IP4 in the cytosol / cellular response to calcium ion / response to calcium ion / small GTPase binding / dendritic spine / cytoskeleton / calmodulin binding / glutamatergic synapse / signal transduction / ATP binding / nucleus / cytosol / cytoplasmSimilarity search - Function Inositol polyphosphate kinase / Inositol polyphosphate kinase / Inositol polyphosphate kinase superfamily / Inositol polyphosphate kinase / D-amino Acid Aminotransferase; Chain A, domain 1 / 2-Layer Sandwich / Alpha BetaSimilarity search - Domain/homology PHOSPHOAMINOPHOSPHONIC ACID-ADENYLATE ESTER / D-MYO-INOSITOL-1,4,5-TRIPHOSPHATE / : / Inositol-trisphosphate 3-kinase ASimilarity search - Component |
|---|
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 1.95 Å X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 1.95 Å |
|---|
 Authors Authors | Gonzalez, B. / Schell, M.J. / Irvine, R.F. / Williams, R.L. |
|---|
 Citation Citation | Journal: Mol.Cell / Year: 2004
Title: Structure of a Human Inositol 1,4,5-Trisphosphate 3-Kinase; Substrate Binding Reveals Why It is not a Phosphoinositide 3-Kinase
Authors: Gonzalez, B. / Schell, M.J. / Letcher, A.J. / Veprintsev, D.B. / Irvine, R.F. / Williams, R.L. |
|---|
| History | | Deposition | Jul 1, 2004 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Sep 9, 2004 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jul 13, 2011 | Group: Advisory / Version format compliance |
|---|
| Revision 1.2 | Jan 24, 2018 | Group: Source and taxonomy / Category: entity_src_gen
Item: _entity_src_gen.pdbx_host_org_ncbi_taxonomy_id / _entity_src_gen.pdbx_host_org_scientific_name ..._entity_src_gen.pdbx_host_org_ncbi_taxonomy_id / _entity_src_gen.pdbx_host_org_scientific_name / _entity_src_gen.pdbx_host_org_strain / _entity_src_gen.pdbx_host_org_variant |
|---|
| Revision 1.3 | May 8, 2024 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Other
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_struct_conn_angle / struct_conn
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj













 Assembly
Assembly




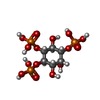



 Mass: 506.196 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N6O12P3 / Comment: AMP-PNP, energy-carrying molecule analogue*YM
Mass: 506.196 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N6O12P3 / Comment: AMP-PNP, energy-carrying molecule analogue*YM Mass: 420.096 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H15O15P3
Mass: 420.096 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H15O15P3 Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn
Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Sample preparation
Sample preparation / Beamline: ID14-4 / Wavelength: 0.9795
/ Beamline: ID14-4 / Wavelength: 0.9795  Processing
Processing