 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1utm: Trypsin specificity as elucidated by LIE calculations, X-ray stru... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1utm | ||||||
|---|---|---|---|---|---|---|---|
| Title | Trypsin specificity as elucidated by LIE calculations, X-ray structures and association constant measurements | ||||||
 Components Components | TRYPSIN I | ||||||
 Keywords Keywords | HYDROLASE / TRYPSIN / INHIBITOR SPECIFICITY / ELECTROSTATIC INTERACTIONS / COLD-ADAPTATION / MOLECULAR DYNAMICS / BINDING FREE ENERGY | ||||||
| Function / homology |  Function and homology information Function and homology informationtrypsin / digestion / serine-type endopeptidase activity / proteolysis / : / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 1.5 Å | ||||||
 Authors Authors | Leiros, H.-K.S. / Brandsdal, B.O. / Andersen, O.A. / Os, V. / Leiros, I. / Helland, R. / Otlewski, J. / Willassen, N.P. / Smalas, A.O. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2004 Title: Trypsin Specificity as Elucidated by Lie Calculations, X-Ray Structures, and Association Constant Measurements Authors: Leiros, H.-K.S. / Brandsdal, B.O. / Andersen, O.A. / Os, V. / Leiros, I. / Helland, R. / Otlewski, J. / Willassen, N.P. / Smalas, A.O. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 6-STRANDED BARREL THIS IS REPRESENTED BY A 7-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1utm.cif.gz | 59.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1utm.ent.gz | 41.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1utm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ut/1utmftp://data.pdbj.org/pub/pdb/validation_reports/ut/1utm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1utjC  1utkC  1utlC  1utnC  1utoC  1utpC  1utqC C: citing same article ( |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 25998.297 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Chemical | ChemComp-PEA / 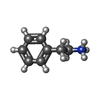  Mass: 122.188 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H12N Mass: 122.188 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H12N |
| #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 109 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 109 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.97 Å3/Da / Density % sol: 37.67 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6 / Details: pH 6.00 | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 37 ℃ / pH: 6 / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 295 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X31 / Wavelength: 1.07 / Beamline: X31 / Wavelength: 1.07 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.07 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→8 Å / Num. obs: 32883 / % possible obs: 97.9 % / Redundancy: 3.39 % / Biso Wilson estimate: 14.25 Å2 / Rmerge(I) obs: 0.048 / Net I/σ(I): 10.1 |
| Reflection shell | Rmerge(I) obs: 0.271 / Mean I/σ(I) obs: 2.8 / % possible all: 93.4 |
| Reflection | *PLUS Highest resolution: 1.5 Å / Num. measured all: 111508 / Rmerge(I) obs: 0.048 |
| Reflection shell | *PLUS % possible obs: 93.4 % / Rmerge(I) obs: 0.271 / Mean I/σ(I) obs: 2.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 1.5→8 Å / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.68 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.17 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.5 Å / Lowest resolution: 8 Å / % reflection Rfree: 7 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
