 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
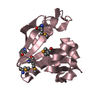
Yorodumi- PDB-1usn: CRYSTAL STRUCTURE OF THE CATALYTIC DOMAIN OF HUMAN FIBROBLAST STR... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1usn | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE CATALYTIC DOMAIN OF HUMAN FIBROBLAST STROMELYSIN-1 INHIBITED WITH THIADIAZOLE INHIBITOR PNU-142372 | ||||||
 Components Components | STROMELYSIN-1 | ||||||
 Keywords Keywords | HYDROLASE / METALLOPROTEASE / FIBROBLAST / COLLAGEN DEGRADATION | ||||||
| Function / homology |  Function and homology information Function and homology informationstromelysin 1 / cellular response to UV-A / regulation of neuroinflammatory response / Assembly of collagen fibrils and other multimeric structures / Activation of Matrix Metalloproteinases / response to amyloid-beta / Collagen degradation / collagen catabolic process / negative regulation of reactive oxygen species metabolic process / extracellular matrix disassembly ...stromelysin 1 / cellular response to UV-A / regulation of neuroinflammatory response / Assembly of collagen fibrils and other multimeric structures / Activation of Matrix Metalloproteinases / response to amyloid-beta / Collagen degradation / collagen catabolic process / negative regulation of reactive oxygen species metabolic process / extracellular matrix disassembly / Degradation of the extracellular matrix / extracellular matrix organization / cellular response to nitric oxide / EGFR Transactivation by Gastrin / regulation of cell migration / negative regulation of phosphatidylinositol 3-kinase/protein kinase B signal transduction / protein catabolic process / cellular response to reactive oxygen species / cellular response to amino acid stimulus / positive regulation of protein-containing complex assembly / metalloendopeptidase activity / metallopeptidase activity / peptidase activity / extracellular matrix / cellular response to lipopolysaccharide / Interleukin-4 and Interleukin-13 signaling / endopeptidase activity / Extra-nuclear estrogen signaling / serine-type endopeptidase activity / innate immune response / mitochondrion / proteolysis / : / extracellular region / zinc ion binding / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Finzel, B.C. / Bryant Junior, G.L. / Baldwin, E.T. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 1998 Title: Structural characterizations of nonpeptidic thiadiazole inhibitors of matrix metalloproteinases reveal the basis for stromelysin selectivity. Authors: Finzel, B.C. / Baldwin, E.T. / Bryant Jr., G.L. / Hess, G.F. / Wilks, J.W. / Trepod, C.M. / Mott, J.E. / Marshall, V.P. / Petzold, G.L. / Poorman, R.A. / O'Sullivan, T.J. / Schostarez, H.J. / Mitchell, M.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1usn.cif.gz | 53.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1usn.ent.gz | 37.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1usn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/us/1usnftp://data.pdbj.org/pub/pdb/validation_reports/us/1usn | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2usnC  1slnS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| 2 | 
| ||||||||||||
| 3 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 18595.684 Da / Num. of mol.: 1 / Fragment: CATALYTIC DOMAIN RESIDUES 83 - 247 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cell: FIBROBLAST / Species (production host): Escherichia coli / Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn#3: Chemical |   Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca#4: Chemical | ChemComp-IN9 / | 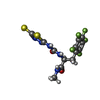  Mass: 427.373 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H10F5N5O2S2 Mass: 427.373 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H10F5N5O2S2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 111 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 111 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.58 Å3/Da / Density % sol: 52.38 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 7 Details: HANGING DROP VAPOR DIFFUSION., pH 7.0, vapor diffusion - hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: SIEMENS / Wavelength: 1.5418 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: Nov 15, 1995 |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→20 Å / Num. obs: 20522 / % possible obs: 95 % / Observed criterion σ(I): 1 / Redundancy: 4.69 % / Rsym value: 0.075 / Net I/σ(I): 10.4 |
| Reflection shell | Resolution: 1.61→1.71 Å / Mean I/σ(I) obs: 1.83 / Rsym value: 0.239 / % possible all: 49 |
| Reflection | *PLUS Num. measured all: 96250 / Rmerge(I) obs: 0.075 |
| Reflection shell | *PLUS % possible obs: 49 % / Rmerge(I) obs: 0.239 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PROTEIN COMPONENT OF STROMELYSIN-1 MODEL (J. W. BECKER, ET AL.; 1SLN) Resolution: 1.8→10 Å / σ(F): 2 Details: SEILECKI ET AL (JMB V134, 1979) PARAMETERS USED IN REFINEMENT. THROUGHOUT THE MODEL, RESIDUES ARE NUMBERED RELATIVE TO THE SEQUENCE OF THE PROENZYME. THE FIRST RESIDUE OF THE MATURE ACTIVE ...Details: SEILECKI ET AL (JMB V134, 1979) PARAMETERS USED IN REFINEMENT. THROUGHOUT THE MODEL, RESIDUES ARE NUMBERED RELATIVE TO THE SEQUENCE OF THE PROENZYME. THE FIRST RESIDUE OF THE MATURE ACTIVE CATALYTIC DOMAIN IS PHE 83. RESIDUES 190-191 ARE PARTIALY DISORDERED. THE CONFORMATION OF THESE ATOMS IS UNCERTAIN. SIDECHAINS OF RESIDUES ASP 111, ASN 214 AND GLU 216 ARE MODELED IN TWO DISCRETE CONFORMATIONS. THE MODEL INCLUDES A CATALYTIC ZN+2 (257), A STRUCTURAL ZN+2 (258), AND THREE BOUND CA+2 IONS (259-261). A THIRD ZN+2 HAS BEEN IDENTIFIED IN THIS CRYSTAL FORM AND RESTS ON THE CRYSTALLOGRAPHIC THREE-FOLD AXIS, COORDINATED BY THREE CRYSTALLOGRAPHICALLY RELATED OCCURANCES OF HIS 96 NE2 AND AN UNKNOWN FOURTH LIGAND. THE FOURTH LIGAND IS MODELED WITH TWO ATOMS. ONE (HOH 263 O) COORDINATED TO ZN 262 ALSO SITS ON THE THREE-FOLD AXIS AND ONE ATOM (HOH 264 O) BONDED (2.05 ANGSTROMS) TO IT. THIS SECOND ATOM IS REPLICATED THREE TIMES BY SYMMETRY. THE FOURTH LIGAND THEREFORE RESEMBLES A CARBONATE OR NITRITE ION.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.11 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROFFT / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 1.9 Å |
