- PDB-1sue: SUBTILISIN BPN' FROM BACILLUS AMYLOLIQUEFACIENS, MUTANT -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 1sue
Title
SUBTILISIN BPN' FROM BACILLUS AMYLOLIQUEFACIENS, MUTANT
Components
SUBTILISIN BPN'
Keywords
SERINE PROTEASE / HYDROLASE
Function / homology
Function and homology information
subtilisin / sporulation resulting in formation of a cellular spore / fibrinolysis / serine-type endopeptidase activity / proteolysis / extracellular region / metal ion binding Similarity search - Function
Resolution: 1.8→1.9 Å / Redundancy: 1.1 % / Rmerge(I) obs: 0.11 / Mean I/σ(I) obs: 2 / Rsym value: 0.11 / % possible all: 60
Reflection shell
*PLUS
% possible obs: 60 %
-
Processing
Software
Name
Version
Classification
XENGEN
datacollection
XENGEN
datareduction
AMoRE
phasing
TNT
5E
refinement
XENGEN
datascaling
Refinement
Method to determine structure: molecular replacement Starting model: REFINED SAME MUTANT IN OTHER CRYSTAL FORM Resolution: 1.8→16 Å / Isotropic thermal model: TNT BCORREL V1.0 / σ(F): 0 / Stereochemistry target values: TNT PROTGEO Details: CRYSTAL MORPHOLOGY VARIES DRAMATICALLY AS A FUNCTION OF IONIC STRENGTH (ADDED SALTS IN 0-500 MM RANGE). OMITTING RESIDUE NUMBERS 75-83 REFLECTS THE ENGINEERED DELETION AND PRESERVES WILD ...Details: CRYSTAL MORPHOLOGY VARIES DRAMATICALLY AS A FUNCTION OF IONIC STRENGTH (ADDED SALTS IN 0-500 MM RANGE). OMITTING RESIDUE NUMBERS 75-83 REFLECTS THE ENGINEERED DELETION AND PRESERVES WILD TYPE NUMBERING OF RESIDUES 84-275. THE ACTIVE SITE IS THE CATALYTIC TRIAD ASP 32, HIS 64, CYS 221 DESCRIBED IN THE SITE RECORD BELOW. SECONDARY STRUCTURE STRUCTURE ASSIGNMENT IS ACCORDING TO KABSCH AND SANDER (BIOPOLYMERS 22, 2577-2637, 1983). RESIDUE SER 221 IS COVALENTLY LINKED TO THE INHIBITOR DIISOPROPYL FLUOROPHOSPHATE (HET GROUP DFP). ONLY ONE OF THE TWO ISOPROPYL GROUPS IS VISIBLE AND INCLUDED IN THE MODEL. SEE HETATM RECORDS DFP, RESIDUE NUMBER 288. CONECT RECORDS SPECIFY BONDING. THE CALCIUM 'A' SITE HAS BEEN DELETED. RESIDUE NA 297 IS IN THE 'B' CATION-BINDING SITE, MONOVALENT SUBSITE. STABILIZING DISULFIDE 3-206 INTRODUCED BY MUTAGENESIS.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj

 Assembly
Assembly

 Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na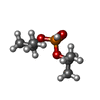
 Mass: 166.155 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15O3P
Mass: 166.155 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15O3P Mass: 18.015 Da / Num. of mol.: 108 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 108 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing