 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qz9: The Three Dimensional Structure of Kynureninase from Pseudomonas ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qz9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | The Three Dimensional Structure of Kynureninase from Pseudomonas fluorescens | ||||||
 Components Components | KYNURENINASE | ||||||
 Keywords Keywords | HYDROLASE / kynurenine / tryptophan / PLP / vitamin B6 / pyridoxal-5'-phosphate | ||||||
| Function / homology |  Function and homology information Function and homology informationkynureninase / kynureninase activity / : / : / quinolinate biosynthetic process / : / NAD+ biosynthetic process / pyridoxal phosphate binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  Pseudomonas fluorescens (bacteria) Pseudomonas fluorescens (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MIR, molecular replacement / Resolution: 1.85 Å X-RAY DIFFRACTION / MIR, molecular replacement / Resolution: 1.85 Å | ||||||
 Authors Authors | Momany, C. / Levdikov, V. / Blagova, L. / Lima, S. / Phillips, R.S. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2004 Title: Three-Dimensional Structure of Kynureninase from Pseudomonas fluorescens. Authors: Momany, C. / Levdikov, V. / Blagova, L. / Lima, S. / Phillips, R.S. | ||||||
| History |
| ||||||
| Remark 600 | HETEROGEN THE LIGAND P3G IS A POLYETHYLENE GLYCOL POLYMER (POSSIBLY MYRISTIC ACID). | ||||||
| Remark 999 | SEQUENCE NO SEQUENCE DATABASE REFERENCE WAS AVAILABLE FOR THIS PROTEIN AT THE TIME OF PROCESSING. ...SEQUENCE NO SEQUENCE DATABASE REFERENCE WAS AVAILABLE FOR THIS PROTEIN AT THE TIME OF PROCESSING. THE SEQUENCE IS DESCRIBED IN CLONING, SEQUENCE, AND EXPRESSION OF KYNURENINASE FROM PSEUDOMONAS FLUORESCENS. KOUSHIK SV, SUNDARARAJU B, MCGRAW RA, PHILLIPS RS. ARCH BIOCHEM BIOPHYS. 1997, V.344, PGS.301-8. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qz9.cif.gz | 168.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qz9.ent.gz | 132.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1qz9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qz/1qz9ftp://data.pdbj.org/pub/pdb/validation_reports/qz/1qz9 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1c0nS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological unit is a dimer generated by applying the symmetry operator (X-Y,-Y,2/3-Z) translated by 1 unit along the b axis |
-Components
| #1: Protein | Mass: 45949.977 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas fluorescens (bacteria) / Plasmid: pTZ18U / Species (production host): Escherichia coli / Production host: |
|---|---|
| #2: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl |
| #3: Chemical | ChemComp-PLP /   Mass: 247.142 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 247.142 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10NO6P |
| #4: Chemical | ChemComp-P3G / 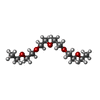  Mass: 250.332 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H26O5 Mass: 250.332 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H26O5 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 428 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 428 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.01 Å3/Da / Density % sol: 38.74 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6 Details: lithium chloride, PEG 400, potassium phosphate, pH 6.0, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
| Crystal grow | *PLUS Method: vapor diffusion, hanging dropDetails: Jancarik, J., (1991) J. Appl. Crystallogr., 24, 409. |
| Components of the solutions | *PLUS Conc.: 10 mg/ml / Common name: protein |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: May 15, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.85→58.72 Å / Num. obs: 29707 / % possible obs: 96.8 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 13.9 % / Biso Wilson estimate: 44 Å2 / Rmerge(I) obs: 0.063 / Net I/σ(I): 25.4 |
| Reflection shell | Resolution: 1.85→1.88 Å / Rmerge(I) obs: 0.608 / Mean I/σ(I) obs: 2 / Num. unique all: 1465 / % possible all: 92.9 |
| Reflection | *PLUS Lowest resolution: 58.7 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR, molecular replacement Starting model: PDB ENTRY 1C0N Resolution: 1.85→19.65 Å / Cor.coef. Fo:Fc: 0.974 / Cor.coef. Fo:Fc free: 0.959 / SU B: 3.057 / SU ML: 0.091 / TLS residual ADP flag: LIKELY RESIDUAL / Isotropic thermal model: ISOTROPIC REFINEMENT WITH TLS / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R: 0.142 / ESU R Free: 0.127 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.478 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.85→19.65 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.85→1.898 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 58.7 Å / % reflection Rfree: 5 % / Rfactor Rfree: 0.192 / Rfactor Rwork: 0.155 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.85 Å / Lowest resolution: 1.9 Å |
