 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1q2n: REFINED Solution NMR structure of the Z domain of STAPHYLOCOCCAL ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1q2n | ||||||
|---|---|---|---|---|---|---|---|
| Title | REFINED Solution NMR structure of the Z domain of STAPHYLOCOCCAL PROTEIN A | ||||||
 Components Components | IMMUNOGLOBULIN G BINDING PROTEIN A | ||||||
 Keywords Keywords | IMMUNE SYSTEM / IMMUNOGLOBULIN-BINDING PROTEIN / THREE-HELICAL BUNDLE STRUCTURE / RESIDUAL DIPOLAR COUPLINGS | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) | ||||||
| Method | SOLUTION NMR / SIMULATED ANNEALING WITH RESTRAINED MOLECULAR DYNAMICS, PROTOCAL USED: ANNEAL.INP | ||||||
 Authors Authors | Zheng, D. / Tashiro, M. / Aramini, J.M. / Montelione, G.T. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2004 Title: Validation of helical tilt angles in the solution NMR structure of the Z domain of Staphylococcal protein A by combined analysis of residual dipolar coupling and NOE data. Authors: Zheng, D. / Aramini, J.M. / Montelione, G.T. #1: Journal: Biochemistry / Year: 1996Title: The Mechanism of Binding Staphylococcal Protein a to Immunoglobin G Does not Involve Helix Unwinding Authors: Jendeberg, L. / Tashiro, M. / Tejero, R. / Lyons, B.A. / Uhlen, M. / Montelione, G.T. / Nilsson, B. #2: Journal: Curr.Opin.Struct.Biol. / Year: 1995Title: Structures of Bacterial Immunoglobulin-Binding Domains and Their Complexes with Immunoglobulin Authors: Tashiro, M. / Montelione, G.T. #3: Journal: Biochemistry / Year: 1993Title: An Improved Strategy for Determining Resonance Assignments for Isotopically Enriched Proteins and its Application to an Engineered Domain of Staphylococcal Protein A Authors: Lyons, B.A. / Tashiro, M. / Cedergren, L. / Nilsson, B. / Montelione, G.T. #4: Journal: J.Mol.Biol. / Year: 1997Title: High Resolution Solution NMR Structure of the Z Domain of Staphylococcal Protein A Authors: Tashiro, M. / Tejero, R. / Zimmerman, D.E. / Celda, B. / Nilsson, B. / Montelione, G.T. | ||||||
| History |
|
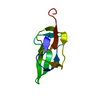
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1q2n.cif.gz | 184.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1q2n.ent.gz | 153.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1q2n.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q2/1q2nftp://data.pdbj.org/pub/pdb/validation_reports/q2/1q2n | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| NMR ensembles |
|
-Components
| #1: Antibody | Mass: 6648.316 Da / Num. of mol.: 1 / Fragment: residues 212-269 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (bacteria) / Cellular location: CELL WALL / Plasmid: PDHZ / Production host: |
|---|
-Experimental details
-Experiment
| Experiment | Method: SOLUTION NMR |
|---|---|
| NMR details | Text: NMR EXPERIMENTS CONDUCTED: 2D PFG-[15N]HSQC, 3D PFG-HNCO, 3D PFG-(HA)CA(CO)NH, 3D PFG-HA(CA)(CO)NH, 3D PFG-HA(CA)NH, 3D PFG-CBCANH, 3D PFG-CBCA(CO)NH, 3D PFG- (HA)CANH, 3D PFG-HN(CA)CO, 3D PFG- ...Text: NMR EXPERIMENTS CONDUCTED: 2D PFG-[15N]HSQC, 3D PFG-HNCO, 3D PFG-(HA)CA(CO)NH, 3D PFG-HA(CA)(CO)NH, 3D PFG-HA(CA)NH, 3D PFG-CBCANH, 3D PFG-CBCA(CO)NH, 3D PFG- (HA)CANH, 3D PFG-HN(CA)CO, 3D PFG-HCCNH-TOCSY, 3D PFG-HCC (CO)NH-TOCSY, 2D [15N]HSQC-IPAP, 3D Ca-coupled HNCO |
- Sample preparation
Sample preparation
| Details | Contents: 1mM Z domain U-15N,13C; 20mM NH4OAc buffer; 95% H2O, 5% D2O. Solvent system: 95% H2O/5% D2O |
|---|---|
| Sample conditions | Ionic strength: 20 mM NH4OAc / pH: 6.5 / Pressure: 1 atm / Temperature: 303.00 K |
| Crystal grow | *PLUS Method: other / Details: NMR |
-NMR measurement
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| NMR spectrometer | Type: Varian MODIFIED UNITY 500 / Manufacturer: Varian / Model: MODIFIED UNITY 500 / Field strength: 500 MHz |
- Processing
Processing
| NMR software |
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method: SIMULATED ANNEALING WITH RESTRAINED MOLECULAR DYNAMICS, PROTOCAL USED: ANNEAL.INP Software ordinal: 1 Details: THE STRUCTURES ARE BASED ON A TOTAL OF 769 CONFORMATIONAL RESTRAINTS. SUMMARY OF EXPERIMENTAL CONSTRAINTS: DISTANCE CONSTRAINTS: TOTAL = 536; INTRA-RESIDUE [I=J] = 224; SEQUENTIAL [(I-J)=1] ...Details: THE STRUCTURES ARE BASED ON A TOTAL OF 769 CONFORMATIONAL RESTRAINTS. SUMMARY OF EXPERIMENTAL CONSTRAINTS: DISTANCE CONSTRAINTS: TOTAL = 536; INTRA-RESIDUE [I=J] = 224; SEQUENTIAL [(I-J)=1] = 142; MEDIUM RANGE [1<(I-J)<5] 105; LONG RANGE [(I-J)>=5] = 65; NUMBER OF DISTANCE CONSTRAINTS RESIDUE = 9.2; DIHEDRAL-ANGLE CONSTRAINTS = 107 RESIDUAL DIPOLAR COUPLING CONSTRAINTS = 126 (34 N-H, 43 HA-CA, 4 TOTAL NUMBER OF CONSTRAINTS PER RESIDUE = 13.3; NUMBER OF LONG RANGE CONSTRAINTS PER RESIDUE = 3.3; NUMBER OF STRUCTURES COMPUTED = 100; NUMBER OF STRUCTURES USED = 10. AVERAGE RESIDUAL CONSTRAINT VIOLATIONS: DISTANCE VIOLATIONS 0.1-0.2 ANG = 7.7, 0. 0.8, >0.5 ANG = 0.3. AVERAGE R.M.S. DISTANCE VIOLATION = 0.03 AN AVERAGE DIHEDRAL ANGLE VIOLATIONS: >0 DEG = 0; RMSD VALUES: BACKBONE ATOMS (N,C,C') = 1.2 ANG; BACKBONE ATOMS(N,C,C') OF ORDERED RESIDUES = 0.4 ANG; ALL HEAVY = 1.7 ANG; ALL HEAVY ATOMS OF ORDERED RESIDUES = 1.0 ANG. PROCHECK USING ORDERED RESIDUES (5-36,39-57): MOST FAVORED REGIONS = 91.4%; ADDITIONAL ALLOWED REGIONS = 8.4%; GENEROUSLY ALLOWED REGIONS = 0.2%; DISALLOWED REGIONS = 0%. | ||||||||||||
| NMR representative | Selection criteria: lowest energy | ||||||||||||
| NMR ensemble | Conformer selection criteria: structures with the lowest energy Conformers calculated total number: 100 / Conformers submitted total number: 10 |
