 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1pvn: The crystal structure of the complex between IMP dehydrogenase ca... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1pvn | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | The crystal structure of the complex between IMP dehydrogenase catalytic domain and a transition state analogue MZP | |||||||||
 Components Components | Inosine-5'-monophosphate dehydrogenase | |||||||||
 Keywords Keywords | OXIDOREDUCTASE / Transition state analogue / IMP dehydrogenase / Mizoribine 5'-monophosphate / distal flap / general base / drug selectivity | |||||||||
| Function / homology |  Function and homology information Function and homology informationIMP dehydrogenase / IMP dehydrogenase activity / GMP biosynthetic process / GTP biosynthetic process / nucleotide binding / protein-containing complex / metal ion binding / identical protein binding / cytoplasm Similarity search - Function | |||||||||
| Biological species |  Tritrichomonas foetus (eukaryote) Tritrichomonas foetus (eukaryote) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | |||||||||
 Authors Authors | Gan, L. / Seyedsayamdost, M. / Shuto, S. / Matsuda, A. / Petsko, G.A. / Hedstrom, L. | |||||||||
 Citation Citation | Journal: Biochemistry / Year: 2003 Title: The Immunosuppressive Agent Mizoribine Monophosphate Forms a Transition State Analogue Complex with Inosine Monophosphate Dehydrogenase Authors: Gan, L. / Seyedsayamdost, M. / Shuto, S. / Matsuda, A. / Petsko, G.A. / Hedstrom, L. | |||||||||
| History |
| |||||||||
| Remark 999 | SEQUENCE IMP dehydrogenase in this structure is a subdomain-deletion mutant. It only contains the ...SEQUENCE IMP dehydrogenase in this structure is a subdomain-deletion mutant. It only contains the catalytic domain with residues 2 to 100 and 227 to 503. Residues 100, 227-230 and 495-503 are disordered in the structure |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1pvn.cif.gz | 310.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1pvn.ent.gz | 247.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1pvn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pv/1pvnftp://data.pdbj.org/pub/pdb/validation_reports/pv/1pvn | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1lrtS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41212.914 Da / Num. of mol.: 4 / Fragment: Catalytic core domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Tritrichomonas foetus (eukaryote) / Gene: IMPDH / Plasmid: pKK233-3 / Production host:  #2: Chemical | ChemComp-K /   Mass: 39.098 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: K#3: Chemical | ChemComp-MZP / 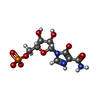  Mass: 337.180 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H12N3O9P Mass: 337.180 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H12N3O9P#4: Chemical | ChemComp-TRS /   Mass: 122.143 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 970 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 970 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.64 Å3/Da / Density % sol: 53.37 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.25 Details: PEG 10,000, MES, Glycerol, KCl, Tris, pH 6.25, VAPOR DIFFUSION, HANGING DROP, temperature 298K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.5 / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-BM-C / Wavelength: 1 Å / Beamline: 14-BM-C / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Sep 23, 2001 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2→30 Å / Num. all: 117771 / Num. obs: 114993 / % possible obs: 99.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 1 / Redundancy: 16 % / Biso Wilson estimate: 11.9 Å2 / Rmerge(I) obs: 0.069 / Net I/σ(I): 22 |
| Reflection shell | Resolution: 2→2.07 Å / Rmerge(I) obs: 0.181 / Mean I/σ(I) obs: 7.8 / Num. unique all: 11629 / % possible all: 99.9 |
| Reflection | *PLUS Num. obs: 113342 / Num. measured all: 1775920 |
| Reflection shell | *PLUS % possible obs: 99.9 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: Tetramer of IMP dehydrogenase, PDB entry 1LRT Resolution: 2→29.71 Å / Rfactor Rfree error: 0.002 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 1 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 52.1099 Å2 / ksol: 0.39474 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.5 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→29.71 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.13 Å / Rfactor Rfree error: 0.006 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Lowest resolution: 8 Å / Num. reflection Rfree: 11341 | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
